DE Analysis
Ha Tran
22/08/2021
Last updated: 2025-11-27
Checks: 7 0
Knit directory: 5_gd_Tcell/1_analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(12345) was run prior to running the
code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version b9f184a. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: .DS_Store
Untracked: figure/scatter2-1.png
Untracked: figure/scatter3-1.png
Untracked: figure/scatter4-1.png
Untracked: figure/scatter5-1.png
Untracked: figure/scatter_3d4-1.png
Untracked: figure/scatter_interactive4-1.png
Untracked: figure/treemap2-1.png
Untracked: figure/treemap3-1.png
Untracked: figure/treemap4-1.png
Untracked: figure/treemap5-1.png
Unstaged changes:
Modified: 0_data/RDS_plots/go_combined_dotPlot.rds
Modified: 0_data/RDS_plots/go_combined_parTerm_dotPlot.rds
Modified: 0_data/RDS_plots/go_dotPlot.rds
Modified: 0_data/RDS_plots/go_parTerm_dotPlot.rds
Modified: 0_data/RDS_plots/go_parTerm_scatter.rds
Modified: 0_data/RDS_plots/kegg_dotPlot.rds
Modified: 0_data/RDS_plots/kegg_path_Hmap.rds
Modified: 0_data/RDS_plots/ma_plots.rds
Modified: 0_data/RDS_plots/react_combined_dotPlot.rds
Modified: 0_data/RDS_plots/react_dotPlot.rds
Modified: 0_data/RDS_plots/vol_plots.rds
Modified: 2_plots/1_QC/PC1_PC2.svg
Modified: 2_plots/1_QC/PC1_PC3.svg
Modified: 2_plots/1_QC/PC2_PC3.svg
Modified: 2_plots/2_DE/heat_down_INT vs CONT.svg
Modified: 2_plots/2_DE/heat_down_INT vs SVX_VAS.svg
Modified: 2_plots/2_DE/heat_down_INT vs VAS.svg
Modified: 2_plots/2_DE/heat_down_SVX vs SVX_VAS.svg
Modified: 2_plots/2_DE/heat_down_SVX_VAS vs CONT.svg
Modified: 2_plots/2_DE/heat_down_VAS vs SVX_VAS.svg
Modified: 2_plots/2_DE/heat_up_INT vs CONT.svg
Modified: 2_plots/2_DE/heat_up_INT vs SVX_VAS.svg
Modified: 2_plots/2_DE/heat_up_INT vs VAS.svg
Modified: 2_plots/2_DE/heat_up_SVX vs SVX_VAS.svg
Modified: 2_plots/2_DE/heat_up_SVX_VAS vs CONT.svg
Modified: 2_plots/2_DE/heat_up_VAS vs SVX_VAS.svg
Modified: 2_plots/2_DE/hist_INT vs CONT.svg
Modified: 2_plots/2_DE/hist_INT vs SVX_VAS.svg
Modified: 2_plots/2_DE/hist_INT vs VAS.svg
Modified: 2_plots/2_DE/hist_SVX vs SVX_VAS.svg
Modified: 2_plots/2_DE/hist_SVX_VAS vs CONT.svg
Modified: 2_plots/2_DE/hist_VAS vs SVX_VAS.svg
Modified: 2_plots/3_FA/go/combine_go_dot.svg
Modified: 2_plots/3_FA/go/parTerm_dot_INT vs CONT.svg
Modified: 2_plots/3_FA/go/parTerm_dot_INT vs SVX_VAS.svg
Modified: 2_plots/3_FA/go/parTerm_dot_SVX vs SVX_VAS.svg
Modified: 2_plots/3_FA/go/parTerm_dot_SVX_VAS vs CONT.svg
Modified: 2_plots/3_FA/go/parTerm_dot_VAS vs SVX_VAS.svg
Modified: 2_plots/3_FA/go/semSim_dendrogram_INT vs CONT.svg
Modified: 2_plots/3_FA/go/semSim_dendrogram_INT vs SVX_VAS.svg
Modified: 2_plots/3_FA/go/semSim_dendrogram_SVX vs SVX_VAS.svg
Modified: 2_plots/3_FA/go/semSim_dendrogram_SVX_VAS vs CONT.svg
Modified: 2_plots/3_FA/go/semSim_dendrogram_VAS vs SVX_VAS.svg
Modified: 2_plots/3_FA/go/semSim_scatter_INT vs CONT.svg
Modified: 2_plots/3_FA/go/semSim_scatter_INT vs SVX_VAS.svg
Modified: 2_plots/3_FA/go/semSim_scatter_SVX vs SVX_VAS.svg
Modified: 2_plots/3_FA/go/semSim_scatter_SVX_VAS vs CONT.svg
Modified: 2_plots/3_FA/kegg/combine_kegg_dot.svg
Modified: 2_plots/3_FA/kegg/heat_Neutrophil extracellular trap formation.svg
Modified: 2_plots/3_FA/kegg/heat_PD-L1 expression and PD-1 checkpoint pathway in cancer.svg
Modified: 2_plots/3_FA/kegg/heat_T cell receptor signaling pathway.svg
Modified: 2_plots/3_FA/kegg/heat_Th1 and Th2 cell differentiation.svg
Modified: 2_plots/3_FA/kegg/heat_Th17 cell differentiation.svg
Modified: 2_plots/3_FA/kegg/kegg_dot_INT vs CONT.svg
Modified: 2_plots/3_FA/kegg/kegg_dot_INT vs SVX_VAS.svg
Modified: 2_plots/3_FA/kegg/kegg_dot_SVX_VAS vs CONT.svg
Modified: 2_plots/3_FA/kegg/kegg_dot_VAS vs SVX_VAS.svg
Modified: 2_plots/3_FA/kegg/kegg_upset_INT vs CONT.svg
Modified: 2_plots/3_FA/kegg/kegg_upset_INT vs SVX_VAS.svg
Modified: 2_plots/3_FA/kegg/kegg_upset_SVX_VAS vs CONT.svg
Modified: 2_plots/3_FA/kegg/kegg_upset_VAS vs SVX_VAS.svg
Modified: 2_plots/3_FA/reactome/combine_react_dot.svg
Modified: 2_plots/3_FA/reactome/react_dot_INT vs CONT.svg
Modified: 2_plots/3_FA/reactome/react_dot_INT vs SVX_VAS.svg
Modified: 2_plots/3_FA/reactome/react_dot_INT vs VAS.svg
Modified: 2_plots/3_FA/reactome/react_dot_SVX vs SVX_VAS.svg
Modified: 2_plots/3_FA/reactome/react_dot_SVX_VAS vs CONT.svg
Modified: 2_plots/3_FA/reactome/react_dot_VAS vs SVX_VAS.svg
Modified: 2_plots/3_FA/reactome/react_upset_INT vs CONT.svg
Modified: 2_plots/3_FA/reactome/react_upset_INT vs SVX_VAS.svg
Modified: 2_plots/3_FA/reactome/react_upset_INT vs VAS.svg
Modified: 2_plots/3_FA/reactome/react_upset_SVX vs SVX_VAS.svg
Modified: 2_plots/3_FA/reactome/react_upset_SVX_VAS vs CONT.svg
Modified: 2_plots/3_FA/reactome/react_upset_VAS vs SVX_VAS.svg
Modified: 2_plots/4_paper/subset_degs.svg
Modified: 2_plots/combine_ipa_dot.svg
Modified: 2_plots/dnf_plot.svg
Modified: 2_plots/intVsvxVAS.svg
Modified: 2_plots/upstream_hmap.svg
Modified: 3_output/DEGs.xlsx
Modified: 3_output/Gene Ontology.xlsx
Modified: 3_output/KEGG.xlsx
Modified: 3_output/Reactome.xlsx
Modified: 3_output/deg_all_new.xlsx
Modified: 3_output/deg_sig_new.xlsx
Modified: 3_output/eigenvalues.xlsx
Modified: 3_output/enrichKEGG_sig.xlsx
Modified: 3_output/reactome_all_new.xlsx
Modified: 3_output/reactome_sig_new.xlsx
Modified: 3_output/semSim_GO_sig.xlsx
Modified: README.html
Modified: README.md
Modified: figure/dot2-1.png
Modified: figure/dot3-1.png
Modified: figure/dot4-1.png
Modified: figure/dot5-1.png
Modified: figure/upset2-1.png
Modified: figure/upset3-1.png
Modified: figure/upset4-1.png
Modified: figure/upset5-1.png
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (1_analysis/deAnalysis.Rmd) and HTML
(docs/deAnalysis.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | d519e7f | Ha Tran | 2024-12-03 | Build site. |
| Rmd | 9fc0156 | Ha Tran | 2024-12-03 | workflowr::wflow_publish(here::here("1_analysis/*.Rmd")) |
| html | 5dce909 | Ha Tran | 2024-11-07 | Build site. |
| html | d71eeb4 | Ha Tran | 2024-10-16 | Build site. |
| Rmd | 5f0c7a1 | Ha Tran | 2024-10-16 | workflowr::wflow_publish(here::here("1_analysis/*.Rmd")) |
| html | ae93fcc | tranmanhha135 | 2024-02-02 | Build site. |
| Rmd | fbcdd69 | tranmanhha135 | 2024-02-02 | workflowr::wflow_publish(here::here("1_analysis/*.Rmd")) |
| html | 2c24612 | tranmanhha135 | 2022-10-13 | build website |
| Rmd | 324032b | tranmanhha135 | 2022-10-11 | resize images |
| Rmd | 43675d3 | tranmanhha135 | 2022-10-06 | Add IPA data |
| html | 43675d3 | tranmanhha135 | 2022-10-06 | Add IPA data |
| html | 11a5cf4 | tranmanhha135 | 2022-10-03 | build wedsite |
| Rmd | 1101367 | tranmanhha135 | 2022-10-02 | Completed functional enrichment for all comparison |
| html | 1101367 | tranmanhha135 | 2022-10-02 | Completed functional enrichment for all comparison |
| html | 68585df | tranmanhha135 | 2022-09-20 | build webste |
| Rmd | 192d010 | tranmanhha135 | 2022-09-20 | functional enrichment with new dataset |
| html | 192d010 | tranmanhha135 | 2022-09-20 | functional enrichment with new dataset |
| html | 6b987dc | tranmanhha135 | 2022-09-08 | Build site. |
| Rmd | ec632aa | tranmanhha135 | 2022-09-08 | workflowr::wflow_publish(all = TRUE, republish = TRUE) |
| html | b9caf15 | tranmanhha135 | 2022-09-08 | Build site. |
| html | 72c4c25 | tranmanhha135 | 2022-09-08 | Build site. |
| Rmd | 0df047f | tranmanhha135 | 2022-09-08 | minor changes to build and publish |
| html | 0df047f | tranmanhha135 | 2022-09-08 | minor changes to build and publish |
| Rmd | 959a5df | tranmanhha135 | 2022-09-07 | rewrite after conversation with Jimmy |
| html | 959a5df | tranmanhha135 | 2022-09-07 | rewrite after conversation with Jimmy |
| html | 54e0166 | Ha Manh Tran | 2022-01-01 | Build site. |
| html | 32454d5 | Ha Manh Tran | 2022-01-01 | Build site. |
| Rmd | c667dd0 | Ha Manh Tran | 2022-01-01 | workflowr::wflow_publish(files = here::here(c("1_analysis/index.Rmd", |
| Rmd | 3b268fc | Ha Tran | 2021-12-09 | multiple FC, reactome, big clean up. |
| html | 3b268fc | Ha Tran | 2021-12-09 | multiple FC, reactome, big clean up. |
| html | b409b38 | Ha Manh Tran | 2021-11-30 | Build site. |
| Rmd | d19f8aa | Ha Manh Tran | 2021-11-30 | workflowr::wflow_publish(files = c("1_analysis/index.Rmd", "1_analysis/figure/setUp.Rmd/", |
| html | f4ba25b | Ha Manh Tran | 2021-11-23 | Build site. |
| Rmd | 1d0620e | Ha Manh Tran | 2021-11-23 | wflow_publish(c("1_analysis/index.Rmd", "1_analysis/setUp.Rmd", |
| Rmd | 516f8e9 | Ha Tran | 2021-11-18 | changed treat lfc to from 0.584 to 1 |
| html | 516f8e9 | Ha Tran | 2021-11-18 | changed treat lfc to from 0.584 to 1 |
| Rmd | 8092dd7 | Ha Tran | 2021-11-04 | initial commit |
| html | 8092dd7 | Ha Tran | 2021-11-04 | initial commit |
Data Setup
# working with data
library(readxl)
library(dplyr)
library(magrittr)
library(readr)
library(tibble)
library(reshape2)
library(tidyverse)
library(ComplexHeatmap)
library(scales)
library(plyr)
# Visualisation:
library(kableExtra)
library(ggplot2)
library(grid)
library(pander)
library(cowplot)
library(pheatmap)
library(VennDiagram)
library(DT)
library(patchwork)
library(kableExtra)
library(extrafont)
loadfonts(device = "all")
# Custom ggplot
library(ggplotify)
library(ggpubr)
library(ggrepel)
library(viridis)
# Bioconductor packages:
library(edgeR)
library(limma)
library(Glimma)
library(pandoc)Import DGElist Data
DGElist object containing the raw feature count, sample metadata, and gene metadata, created in the Set Up stage.
# load DGElist previously created in the set up
dge <- readRDS(here::here("0_data/RDS_objects/dge.rds"))
# to increase the knitting speed. change to T to save all plots
savePlots <- T
export <- T# Theme
bossTheme <- readRDS(here::here("0_data/functions/bossTheme.rds"))
bossTheme_bar <- readRDS(here::here("0_data/functions/bossTheme_bar.rds"))
groupColour <- readRDS(here::here("0_data/functions/groupColour.rds"))
groupColour_dark <- readRDS(here::here("0_data/functions/groupColour_dark.rds"))
expressionCol <- readRDS(here::here("0_data/functions/expressionCol.rds"))
expressionCol_dark <- readRDS(here::here("0_data/functions/expressionCol_dark.rds"))
compColour <- readRDS(here::here("0_data/functions/compColour.rds"))
patch <- readRDS(here::here("0_data/functions/patch.rds"))
patch_ymax <- readRDS(here::here("0_data/functions/patch_ymax.rds"))
DT <- readRDS(here::here("0_data/functions/DT.rds"))
# Plotting
convert_to_superscript <- readRDS(here::here("0_data/functions/convert_to_superscript.rds"))
exponent <- readRDS(here::here("0_data/functions/exponent.rds"))
format_y_axis <- readRDS(here::here("0_data/functions/format_y_axis.rds"))Initial Parameterisation
The varying methods used to identify differential expression all rely on similar initial parameters. These include:
The Design Matrix,
Estimation of Dispersion, and
Contrast Matrix
Design Matrix
The experimental design can be parameterised in a one-way layout where one coefficient is assigned to each group. The design matrix formulated below contains the predictors of each sample
# null design with unit vector for generation of voomWithQualityWeights downstream
null_design <- matrix(1, ncol = 1, nrow = ncol(dge))
# setup full design matrix with sample_group
full_design <- model.matrix(~ 0 + group, data = dge$samples)
# remove "sample_group" from each column names
colnames(full_design) <- gsub( "group", "", colnames(full_design))
# display the full_design matrix
full_design %>% as.data.frame() %>% DT(., "Table: Design matrix")colnames(full_design) <- make.names(colnames(full_design))Contrast Matrix
The contrast matrix is required to provide a coefficient to each comparison and later used to test for significant differential expression with each comparison group
contrast <- limma::makeContrasts(
INTvsCONT = INT - CONT,
INTvsSVX_VAS = INT - SVX_VAS,
SVXvsSVX_VAS = SVX - SVX_VAS,
VASvsSVX_VAS = VAS - SVX_VAS,
SVX_VASvsCONT = SVX_VAS - CONT,
INTvsVAS = INT - VAS,
levels = full_design)
colnames(contrast) <- c("INT vs CONT", "INT vs SVX_VAS", "SVX vs SVX_VAS", "VAS vs SVX_VAS", "SVX_VAS vs CONT", "INT vs VAS")
contrast %>% DT(., "Table: Contrast matrix")Limma-Voom
Apply voom transformation
Voom is used to estimate the mean-variance relationship of the data, which is then used to calculate and assign a precision weight for each of the observation (gene). This observational level weights are then used in a linear modelling process to adjust for heteroscedasticity. Log count (logCPM) data typically show a decreasing mean-variance trend with increasing count size (expression).
However, for some dataset with potential sample outliers,
voomWithQualityWeights can be used to calculate
sample-specific quality weights. The application of observational and
sample-specific weights can objectively and systematically correct for
outliers and better than manually removing samples in cases where there
are no clear-cut reasons for replicate variations
Observational-level weights
# voom tranformation without sample weights
voom <- limma::voom(counts = dge, design = full_design, plot = TRUE,)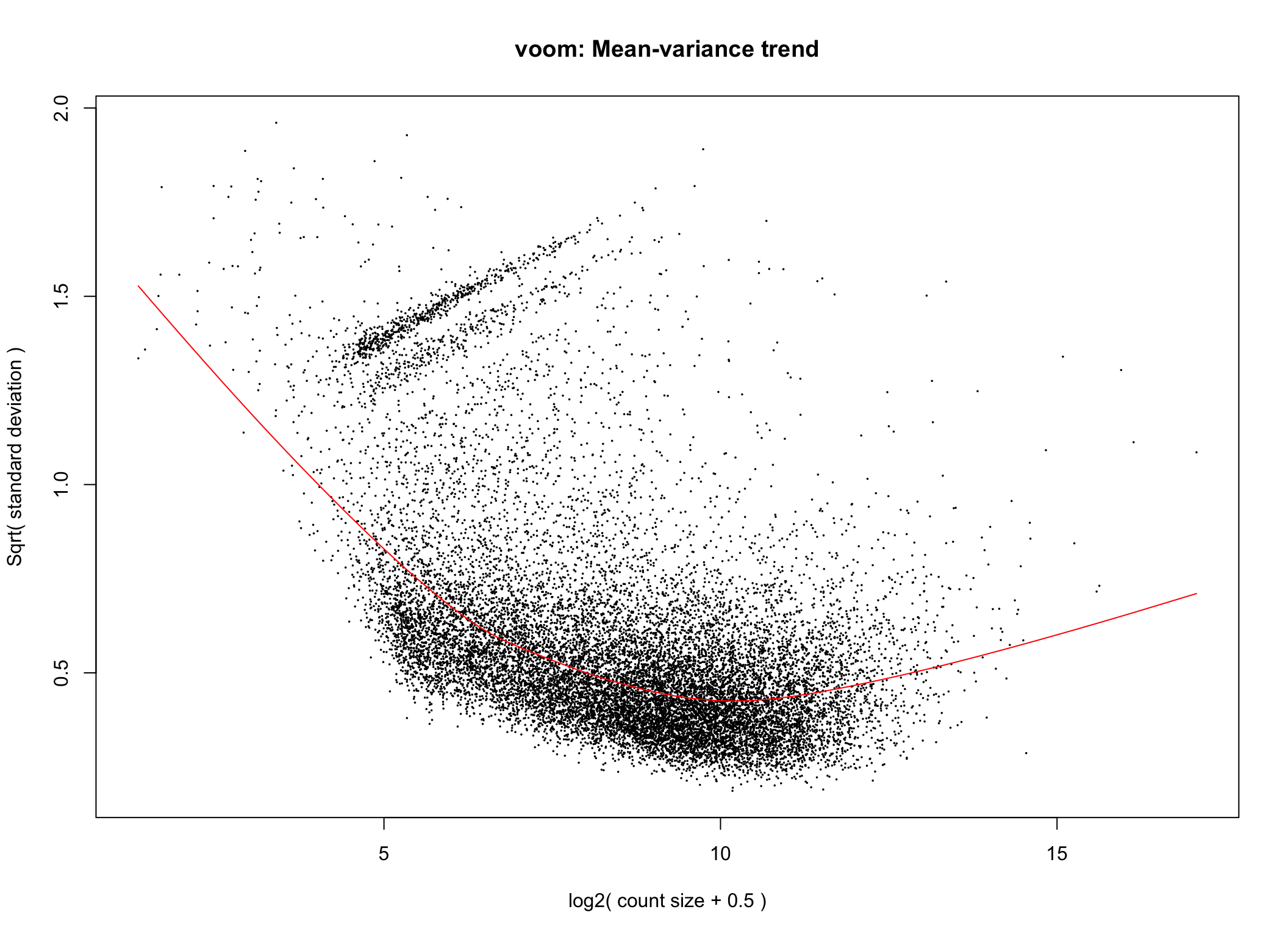
Voom transformation with observational weights
Observational and group level weights
# voom transformation with sample weights using full_design matrix for group-specific weights
voom1 <- limma::voomWithQualityWeights(counts = dge, design = full_design, plot = TRUE)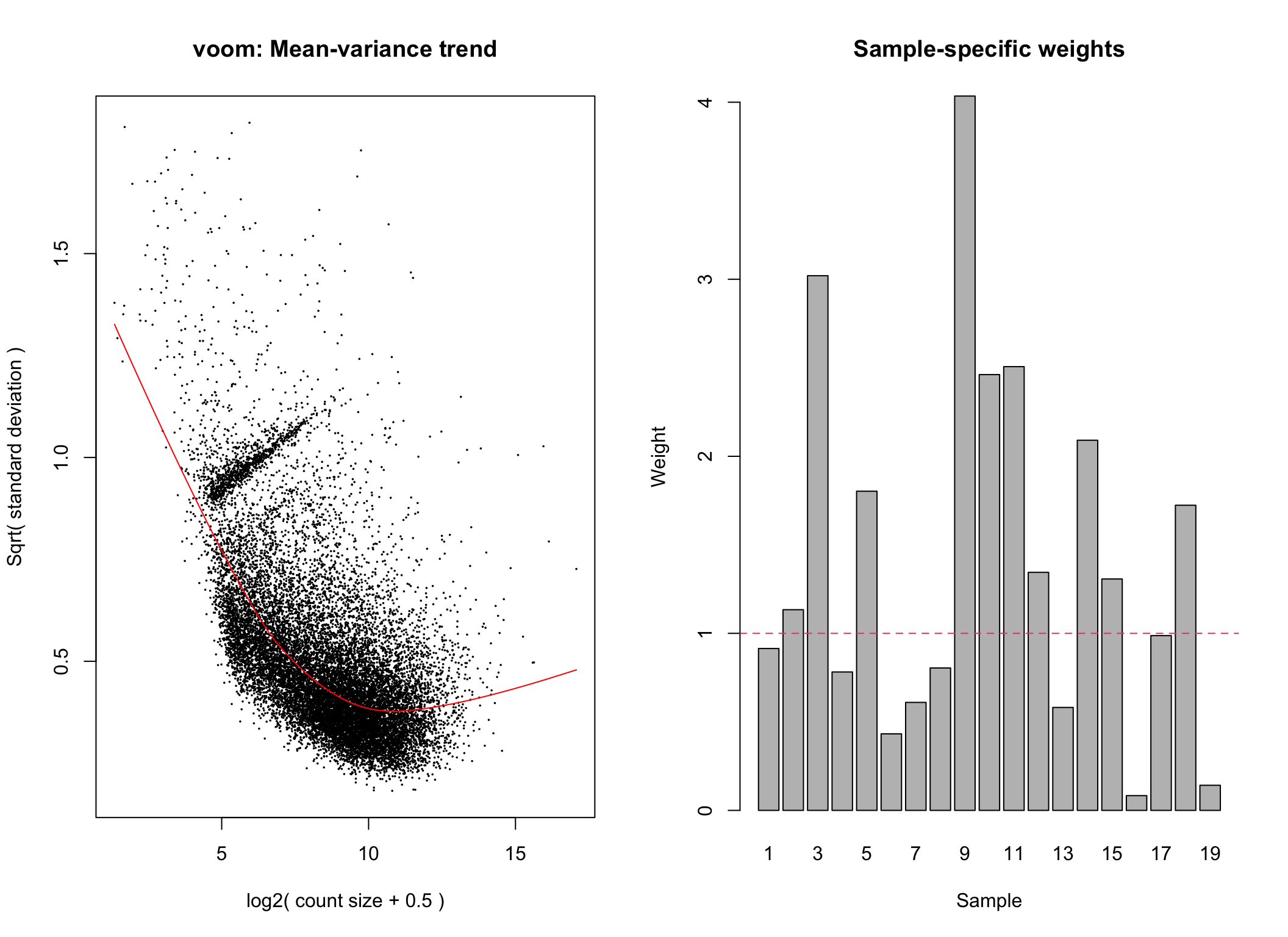
Voom transformation with observational and group-specific weights
Observational and sample level weights
# voom transformation with sample weights using null design matrix
voom2 <- limma::voomWithQualityWeights(counts = dge,design = null_design, plot = TRUE)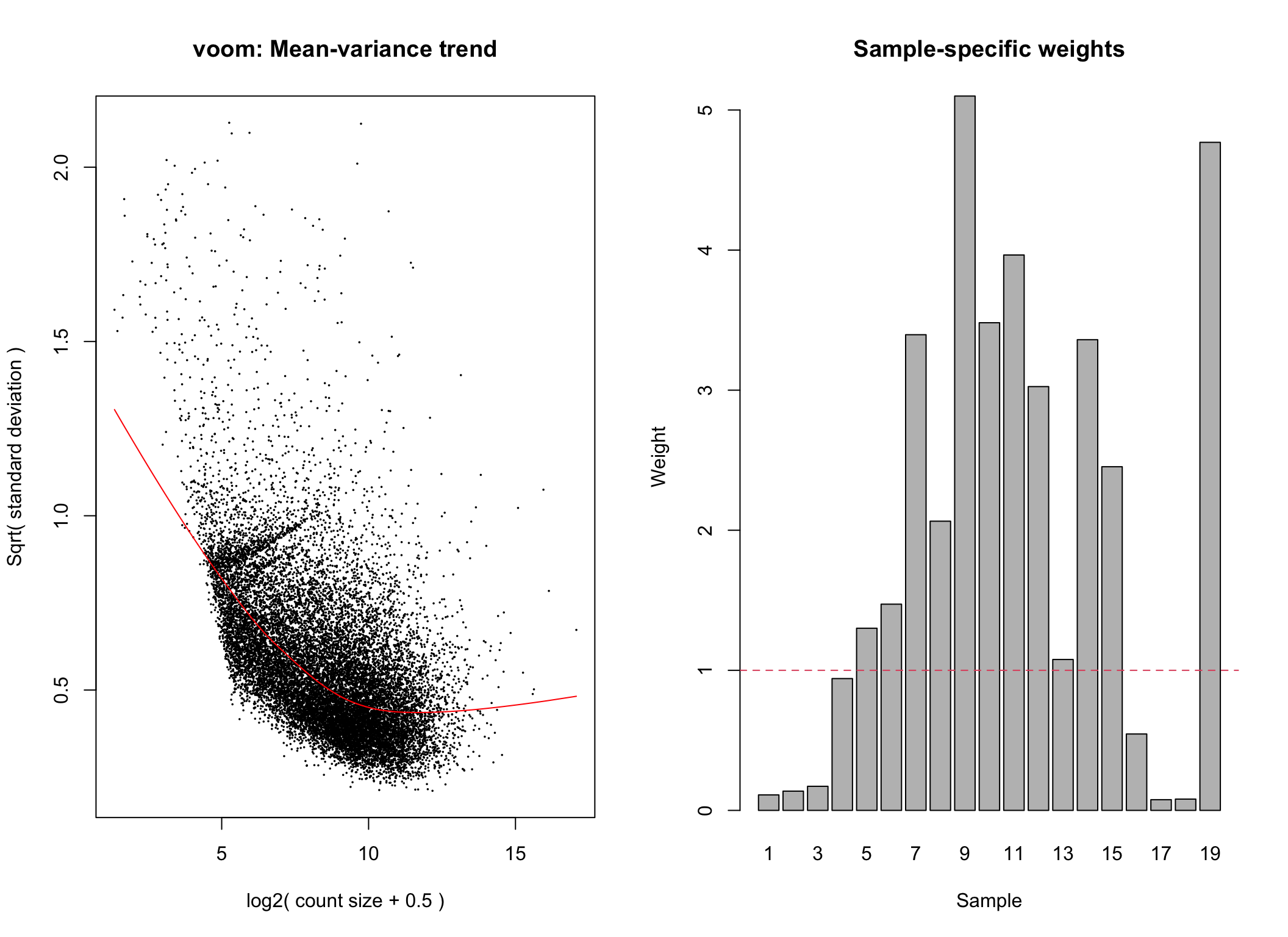
Voom transformation with observational and sample-specific weights
Visualisation of Voom transformation
Groups and sample weights
Equal quality samples should ideally receive a weight of 1. When weights are calculated across all samples irrespective of their groups, the two extreme VAS groups have reduced weights
# function for extracting weights from voom transformation and generating a bar plot
lapply(1:2, function(num){
name <- get(paste0('voom',num))
name$targets %>%
rownames_to_column("Sample") %>%
as_tibble() %>%
ggplot(aes(x = Sample, y = sample.weights, fill = group)) +
scale_fill_manual(values = groupColour) +
geom_bar(stat = "identity", alpha = 0.8) +
labs(x = "", y = "Sample Weights", fill = "") +
geom_hline(yintercept = 1) +
bossTheme()+
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5))
}) %>% patch(., legend = "right") %>% patch_ymax()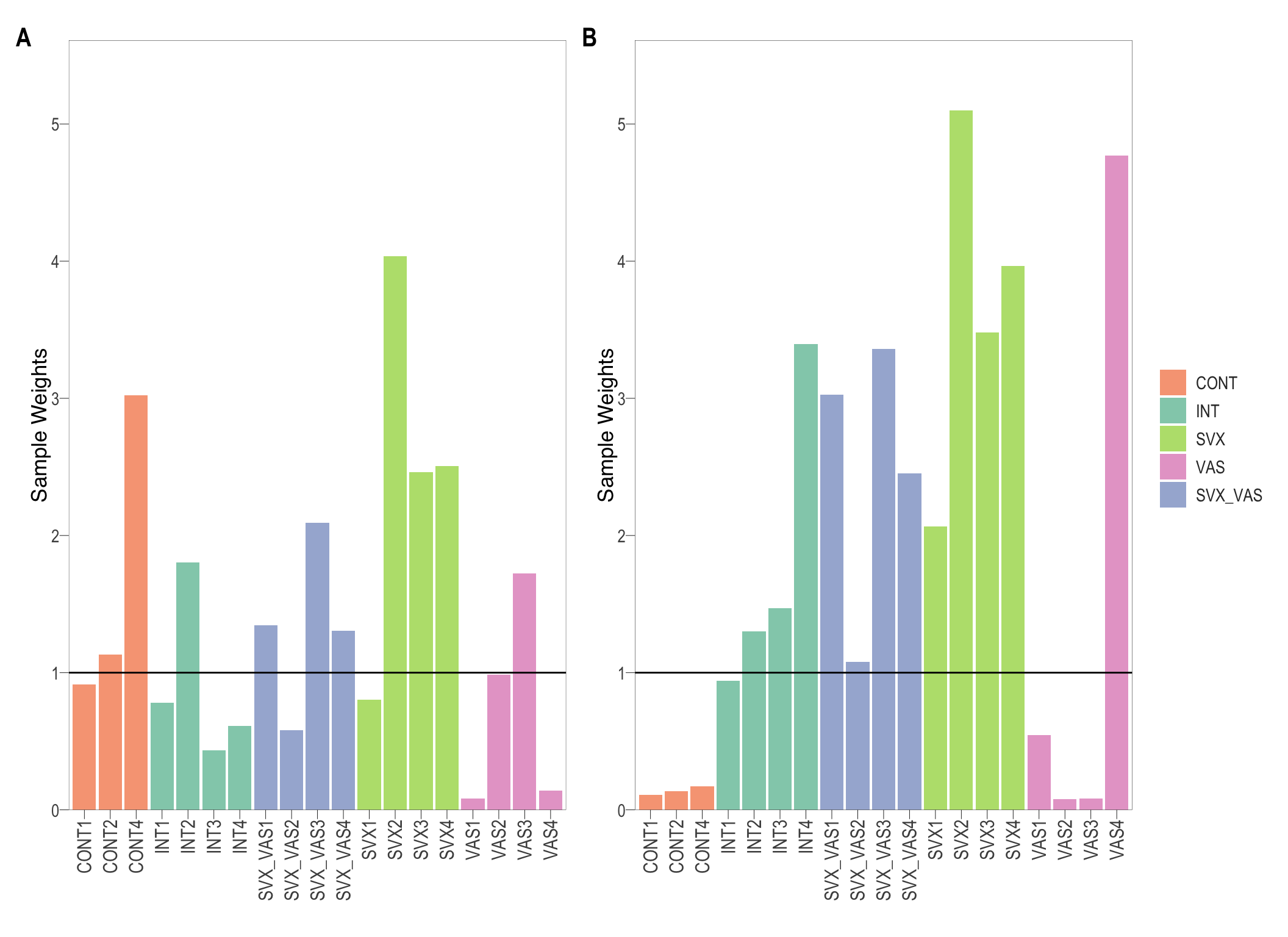
Group and samples specific weights
PCA
After the voom transformation, another PCA plot can be generated. This time the sample weights are represented by size.
# Function for performing pca and generating plots
voom_pca <- function(voom_trans, titleIndex){
title <- c("Group-level weights", "Sample-level weights")
pca <- voom_trans$E %>%
t() %>%
prcomp()
voom_trans$targets <- voom_trans$targets %>% as.data.frame %>% rownames_to_column("Sample_Name")
pca$x %>%
as.data.frame() %>%
rownames_to_column("Sample_Name") %>%
as_tibble() %>%
dplyr::select(Sample_Name, PC1, PC2) %>%
left_join(voom_trans$targets, by = "Sample_Name") %>%
mutate(sample.weights = round(sample.weights, 3)) %>%
ggplot(aes(PC1, PC2, colour = group, size = sample.weights, label = Sample_Name)) +
geom_label_repel(box.padding = grid::unit(0.5,"lines"), size = 3, label.size = 0.15, show.legend = F) +
geom_point(alpha = 0.5) +
scale_color_manual(values = groupColour_dark) +
scale_size_continuous(limits=c(0,5)) +
labs(x = paste0("PC1 (", percent(summary(pca)$importance["Proportion of Variance","PC1"]),")"),
y = paste0("PC2 (", percent(summary(pca)$importance["Proportion of Variance","PC2"]),")"),
colour = "",
size = "Weights",
title = title[titleIndex %>% as.numeric()]) +
bossTheme(legend = "bottom") +
guides(colour = guide_legend(override.aes = list(size = 3)))
}
# iterate with lapply for voom 1 and 2
lapply(c("1","2"), function(y){
voom_pca(voom_trans = get(paste0('voom',y)), titleIndex = y)
}) %>% patch(., legend = "bottom")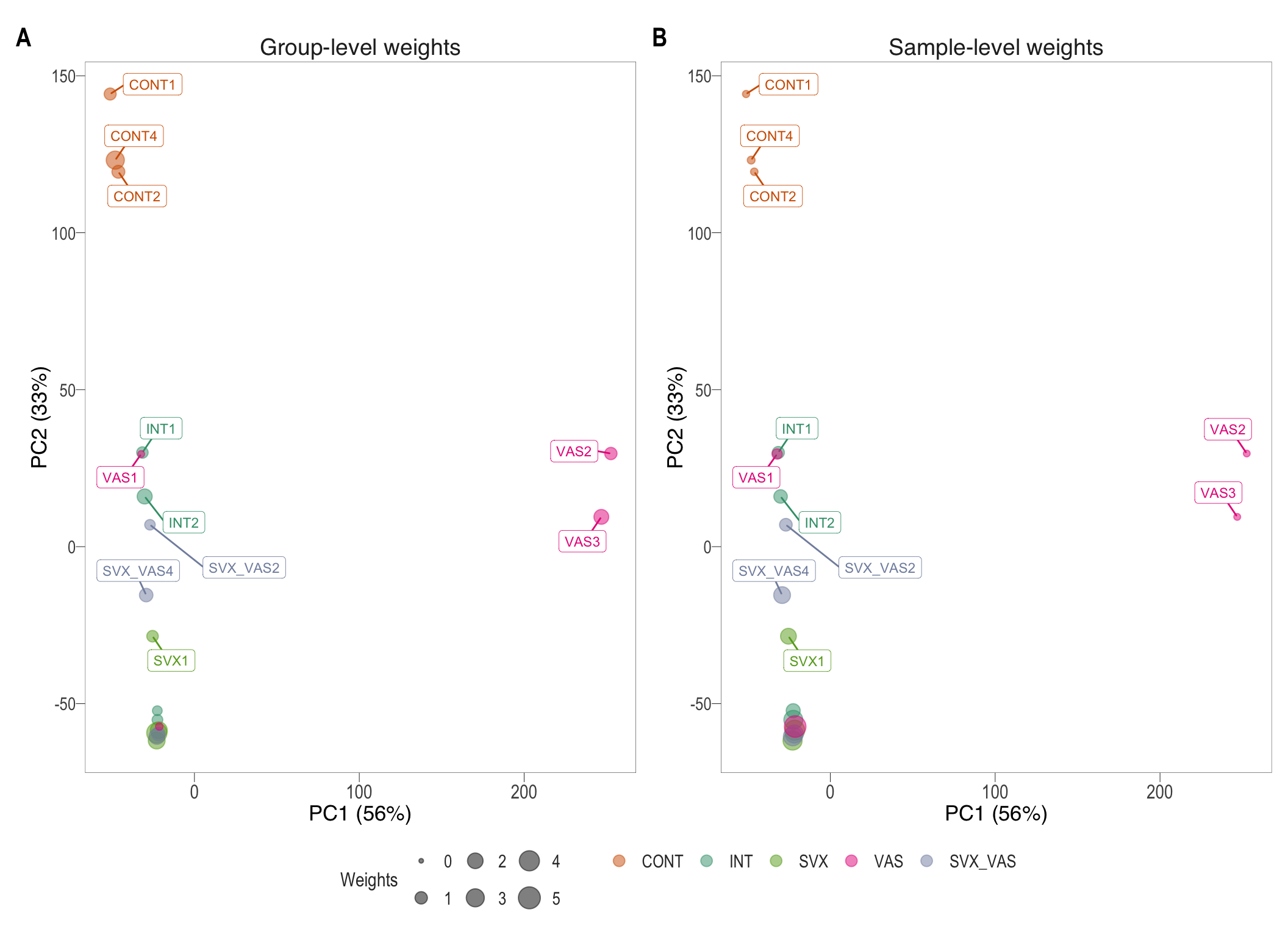
PCA plots after voom quality weight transformations. Group-specific weights(left) and sample-specific weights (right)
PC1 increased and PC2 decreased slightly from the previous PCA plot. The clustering of do not appear to signficantly change, however, the addition of sample weights should give a better fit for linear model. Following voom transformation, the observational and sample-specific weights are used to fit the linear model and
Apply linear model
Without FC cutoff using TREAT After playing around
with the P value adjustment method and the p.value/adj.p.Value cutoff.
The DE analysis of sample-weighted data at an un-adjusted P value of
0.01 generated the most favorable number of DE genes
(Table 1). Adjustment of p-value through any method removes
majority of DE genes from most comparison, even when the adjustment
method is relatively relaxed (e.g. FDR and
BH).
With FC cutoff using TREAT However, without FC
cutoff and p-value of 0.01, the INT vs CONT comparison
still have over 8000 DE genes. Thus, we can afford to be more stringent
with our adjustment method and adj.p.val cutoff. Additionally, when the
list of DE genes is large, we can apply a fold change cutoff through
application of TREAT to prioritise the genes with greater
fold changes and potentially more biologically relevant. Idealy, we are
aiming for ~300 genes genes. IPA analysis with this number of genes
should generate meaningful results.
Importantly, the FC threshold used in TREAT should be
chosen as a small value below which results should be ignored, instead
of a target fold-change. In general, a modest fold-change of 1.1 - 1.5
is recommended. However, it is more important to select a fold-change
cutoff that generates a sufficiently small list of DE genes.
A fold-change value of 1.5 and FDR<0.05, generated a
sufficiently small number of DE genes for the INT vs CONT comparison.
This should be sufficient for functional enrichment analysis (Table
12).
# specifying FC of interest
options(digits = 6)
fc <- c("none", 1.1, 1.2, 1.5)FC=none
# function for applying linear model, generate decideTest table, and extract topTable
limmaFit <- function(x, fc, adjMethod, p.val, tableNum, list = F){
lm <- limma::lmFit(object = x, design = full_design) %>%
contrasts.fit(contrasts = contrast)
if (fc == "none") {
lm <- lm %>% limma::eBayes()
} else {
lm <- lm %>% limma::treat(fc = as.numeric(fc))
}
if (list == TRUE) {
if (fc == "none") {
lm_all <- lapply(1:ncol(lm), function(y){
limma::topTable(lm, coef = y, number = Inf, adjust.method = adjMethod)
})
} else {
lm_all <- lapply(1:ncol(lm), function(y){
limma::topTreat(lm, coef = y, number = Inf, adjust.method = adjMethod)
})
}
lapply(lm_all, function(x) {
df <- x %>% as.data.frame() %>%
dplyr::mutate(expression = case_when(
adj.P.Val <= p.val & logFC >=0 ~ "up",
adj.P.Val <= p.val & logFC <0 ~ "down",
TRUE ~ "insig"))
df$expression <- factor(df$expression, levels = c("insig", "down", "up"))
# df$entrezid <- df$entrezid %>% as.character()
return(df)
})
} else {
lm_dt <- decideTests(object = lm, adjust.method = adjMethod, p.value = p.val)
print(knitr::kable(summary(lm_dt)
, caption = paste0("TABLE ",tableNum, ": Number of significant DE genes from '", deparse(substitute(x)), "' with '", adjMethod, "' adjusment method, and at a p-value/adj.p-value of ", p.val)) %>%
kable_styling(bootstrap_options = c("striped", "hover")))
}
}
limmaFit(x = voom2, fc[1], adjMethod = "none", p.val = 0.01, 1)| INT vs CONT | INT vs SVX_VAS | SVX vs SVX_VAS | VAS vs SVX_VAS | SVX_VAS vs CONT | INT vs VAS | |
|---|---|---|---|---|---|---|
| Down | 305 | 98 | 81 | 96 | 494 | 58 |
| NotSig | 14943 | 16988 | 17028 | 16987 | 14566 | 17116 |
| Up | 1974 | 136 | 113 | 139 | 2162 | 48 |
limmaFit(x = voom2, fc[1], adjMethod = "fdr", p.val = 0.1, 2)| INT vs CONT | INT vs SVX_VAS | SVX vs SVX_VAS | VAS vs SVX_VAS | SVX_VAS vs CONT | INT vs VAS | |
|---|---|---|---|---|---|---|
| Down | 510 | 5 | 0 | 0 | 1021 | 0 |
| NotSig | 14423 | 17213 | 17222 | 17220 | 13522 | 17222 |
| Up | 2289 | 4 | 0 | 2 | 2679 | 0 |
limmaFit(x = voom2, fc[1], adjMethod = "fdr", p.val = 0.05, 3)| INT vs CONT | INT vs SVX_VAS | SVX vs SVX_VAS | VAS vs SVX_VAS | SVX_VAS vs CONT | INT vs VAS | |
|---|---|---|---|---|---|---|
| Down | 134 | 0 | 0 | 0 | 328 | 0 |
| NotSig | 15484 | 17221 | 17222 | 17220 | 15001 | 17222 |
| Up | 1604 | 1 | 0 | 2 | 1893 | 0 |
FC=1.1
limmaFit(x = voom2, fc[2], adjMethod = "none", p.val = 0.01,4)| INT vs CONT | INT vs SVX_VAS | SVX vs SVX_VAS | VAS vs SVX_VAS | SVX_VAS vs CONT | INT vs VAS | |
|---|---|---|---|---|---|---|
| Down | 187 | 40 | 10 | 24 | 344 | 13 |
| NotSig | 15205 | 17126 | 17188 | 17140 | 14883 | 17200 |
| Up | 1830 | 56 | 24 | 58 | 1995 | 9 |
limmaFit(x = voom2, fc[2], adjMethod = "fdr", p.val = 0.1, 5)| INT vs CONT | INT vs SVX_VAS | SVX vs SVX_VAS | VAS vs SVX_VAS | SVX_VAS vs CONT | INT vs VAS | |
|---|---|---|---|---|---|---|
| Down | 253 | 0 | 0 | 0 | 601 | 0 |
| NotSig | 15006 | 17222 | 17222 | 17222 | 14292 | 17222 |
| Up | 1963 | 0 | 0 | 0 | 2329 | 0 |
limmaFit(x = voom2, fc[2], adjMethod = "fdr", p.val = 0.05, 6)| INT vs CONT | INT vs SVX_VAS | SVX vs SVX_VAS | VAS vs SVX_VAS | SVX_VAS vs CONT | INT vs VAS | |
|---|---|---|---|---|---|---|
| Down | 55 | 0 | 0 | 0 | 151 | 0 |
| NotSig | 15775 | 17222 | 17222 | 17222 | 15430 | 17222 |
| Up | 1392 | 0 | 0 | 0 | 1641 | 0 |
FC=1.2
limmaFit(x = voom2, fc[3], adjMethod = "none", p.val = 0.01,7)| INT vs CONT | INT vs SVX_VAS | SVX vs SVX_VAS | VAS vs SVX_VAS | SVX_VAS vs CONT | INT vs VAS | |
|---|---|---|---|---|---|---|
| Down | 80 | 13 | 3 | 10 | 165 | 4 |
| NotSig | 15558 | 17191 | 17215 | 17183 | 15320 | 17213 |
| Up | 1584 | 18 | 4 | 29 | 1737 | 5 |
limmaFit(x = voom2, fc[3], adjMethod = "fdr", p.val = 0.1, 8)| INT vs CONT | INT vs SVX_VAS | SVX vs SVX_VAS | VAS vs SVX_VAS | SVX_VAS vs CONT | INT vs VAS | |
|---|---|---|---|---|---|---|
| Down | 74 | 0 | 0 | 0 | 202 | 0 |
| NotSig | 15588 | 17222 | 17222 | 17222 | 15199 | 17222 |
| Up | 1560 | 0 | 0 | 0 | 1821 | 0 |
limmaFit(x = voom2, fc[3], adjMethod = "fdr", p.val = 0.05, 9)| INT vs CONT | INT vs SVX_VAS | SVX vs SVX_VAS | VAS vs SVX_VAS | SVX_VAS vs CONT | INT vs VAS | |
|---|---|---|---|---|---|---|
| Down | 15 | 0 | 0 | 0 | 47 | 0 |
| NotSig | 16161 | 17222 | 17222 | 17222 | 15924 | 17222 |
| Up | 1046 | 0 | 0 | 0 | 1251 | 0 |
FC=1.5
limmaFit(x = voom2, fc[4], adjMethod = "none", p.val = 0.01,10)| INT vs CONT | INT vs SVX_VAS | SVX vs SVX_VAS | VAS vs SVX_VAS | SVX_VAS vs CONT | INT vs VAS | |
|---|---|---|---|---|---|---|
| Down | 14 | 1 | 0 | 2 | 29 | 0 |
| NotSig | 16238 | 17216 | 17222 | 17214 | 16132 | 17220 |
| Up | 970 | 5 | 0 | 6 | 1061 | 2 |
limmaFit(x = voom2, fc[4], adjMethod = "fdr", p.val = 0.1, 11)| INT vs CONT | INT vs SVX_VAS | SVX vs SVX_VAS | VAS vs SVX_VAS | SVX_VAS vs CONT | INT vs VAS | |
|---|---|---|---|---|---|---|
| Down | 6 | 0 | 0 | 0 | 14 | 0 |
| NotSig | 16542 | 17222 | 17222 | 17222 | 16385 | 17222 |
| Up | 674 | 0 | 0 | 0 | 823 | 0 |
limmaFit(x = voom2, fc[4], adjMethod = "fdr", p.val = 0.05, 12)| INT vs CONT | INT vs SVX_VAS | SVX vs SVX_VAS | VAS vs SVX_VAS | SVX_VAS vs CONT | INT vs VAS | |
|---|---|---|---|---|---|---|
| Down | 2 | 0 | 0 | 0 | 5 | 0 |
| NotSig | 16834 | 17222 | 17222 | 17222 | 16712 | 17222 |
| Up | 386 | 0 | 0 | 0 | 505 | 0 |
Differential Gene Expression analysis
For the first Intact vs Control comparison, a rigorous
statistical test was used to reduce the list of DE genes down to a more
biologically relevant number. This included testing significance
relative to a fold change threshold (TREAT). For this comparison, genes
significantly above of FC of 1.5 and FDR <
0.05 are visualised.
For the other comparisons (Intact vs SVX_VAS,
SVX vs SVX_VAS, and VAS vs SVX_VAS), a less
stringent test was applied to identify significant DE genes while
maintaining sufficient number of downstream functional enrichment
analysis. Genes with P.value < 0.01 are visualised
as follows:
P-value histogram: illustrates the distribution of p-values. As the stringency increases (increasing FC threshold), the distribution shifts towards
1, thus insignificant.MA plot: helps visualise and identify genes with significant changes in expression. Points deviating from the central axis often indicate differentially expressed genes, allowing assessment of the magnitude and consistency of expression changes across conditions.
- \(x-axis =\) average expression, in log counts per million (CPM)
- \(y-axis =\) log fold change between conditions
Volcano plot: shows significantly differentially expressed genes appearing as points that are both statistically significant (located at the top) and have substantial fold changes (located on the left or right sides). This visualization enables identification of genes that are statistically and biologically significant.
- \(x-axis =\) log fold change between conditions
- \(y-axis =\) negative logarithm of the FDR-adjusted p-values
Heatmap: visualize gene expression patterns across different experimental conditions. Rows are genes, columns represent samples, and the colour intensity indicates the expression level of a gene in a specific sample. The genes are also clustered based on similar expression patterns, which provides insights into the overall structure and relationships within large datasets.
- These heatmaps illustrates the top 30 most significant DE genes
Venn diagram: visualises the significant DE gene overlap between the previous RNA-seq experiment and the current.
comp <- colnames(contrast)
options(digits = 6)
# High stringency for the first comparison
highStringency <- limmaFit(x = voom2, fc = 1.5, adjMethod = "fdr", p.val = 0.05, list = T)
highStringency_sig <- lapply(1:ncol(contrast), function(i){filter(highStringency[[i]], adj.P.Val < 0.05)})
# Low stringency for the other three comparison
lowStringency <- limmaFit(x = voom2, fc = "none", adjMethod = "none", p.val = 0.01, list = T)
lowStringency_sig <- lapply(1:ncol(contrast), function(i){filter(lowStringency[[i]], adj.P.Val < 0.01)})
lm_all <- list(highStringency[[1]], lowStringency[[2]], lowStringency[[3]], lowStringency[[4]], highStringency[[5]], lowStringency[[6]]) %>% setNames(colnames(contrast))
lm_sig <- list(highStringency_sig[[1]], lowStringency_sig[[2]], lowStringency_sig[[3]], lowStringency_sig[[4]], highStringency_sig[[5]],lowStringency_sig[[6]]) %>% setNames(colnames(contrast))INT vs CONT
P-val histogram
lm_hist <- list()
for (name in comp) {
lm_hist[[name]] <- ggplot(lm_all[[name]] %>% as.data.frame(), aes(x = P.Value)) +
geom_histogram(bins = 50, fill="#8DA0CB",colour= "white", linewidth = 0.2, alpha=0.9) +
scale_y_continuous(expand = expansion(mult = c(0, .1))) +
labs(x = "P values", y = "Counts") +
bossTheme()
if (savePlots == T) {
ggsave(paste0("hist_",name,".svg"),
plot = lm_hist[[name]],
path = here::here("2_plots/2_DE/"),
width = 13,
height = 11,
units = "cm")
}
}
lm_hist[[1]]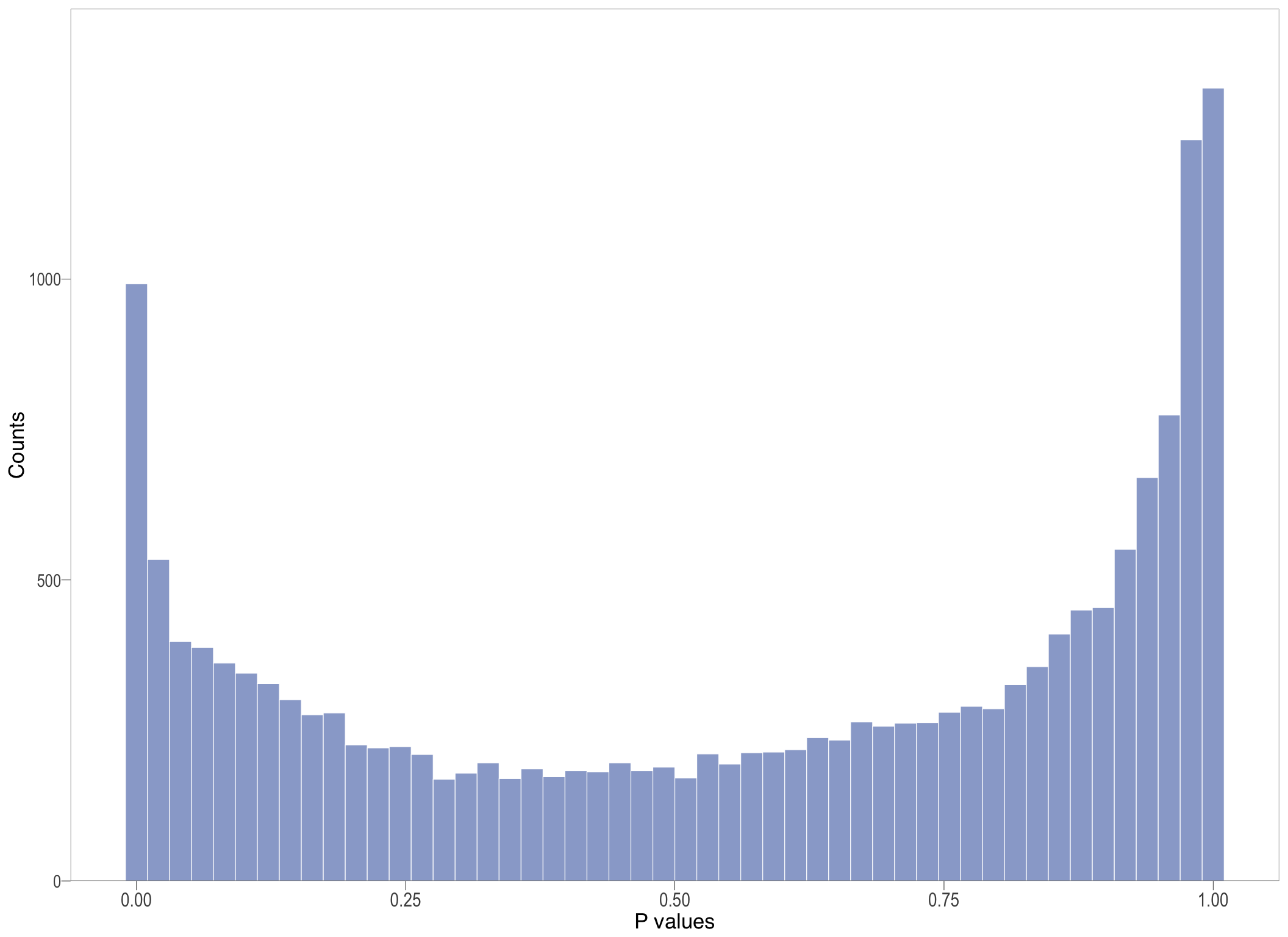
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
MA plot
ma <- list()
for (name in comp) {
top <- 5
top_limma <- bind_rows(
lm_all[[name]] %>%
dplyr::filter(expression == "up") %>%
arrange(adj.P.Val, desc(abs(logFC))) %>%
head(top),
lm_all[[name]] %>%
dplyr::filter(expression == "down") %>%
arrange(adj.P.Val, desc(abs(logFC))) %>%
head(top)
)
invisible(top_limma %>% as.data.frame())
max <- max(lm_all[[name]]$logFC)
ma[[name]] <- lm_all[[name]] %>% dplyr::arrange(expression) %>%
ggplot(aes(x = AveExpr, y = logFC)) +
geom_point(aes(colour = expression, size = expression),show.legend = T, alpha = 0.7, stroke =0) +
geom_label_repel(data = top_limma, # map labels, visit ?geom_label_repel
mapping = aes(label = gene_name),
size = 3,
label.padding = 0.15,
label.size = 0,
label.r = 0.15,
box.padding = 0.9,
point.padding = 0.5,
segment.size = 0.3,
segment.color = "grey50"
) +
labs(
x = expression("log"[2] * "CPM"),
y = expression("log"[2] * "FC"),
colour = "Expression") +
geom_hline(yintercept = 0, linetype = "dashed") +
scale_y_continuous(limits = c(-max,max), expand = expansion(mult = c(0.05, 0.05))) +
scale_size_manual(values = c(1.5,2.4,2.4), guide = "none") +
scale_fill_manual(values = expressionCol) +
scale_color_manual(labels = c(paste0("NS: ", sum(lm_all[[name]]$expression == "insig"), " "),
paste0("Down: ", sum(lm_all[[name]]$expression == "down"), " "),
paste0("Up: ", sum(lm_all[[name]]$expression == "up"), " ")),
values = expressionCol_dark) +
bossTheme(base_size = 14, legend = "bottom") +
guides(colour = guide_legend(override.aes = list(size = 2.4)))
if (savePlots) {
ggsave(paste0("ma_", name, ".png"),
plot = ma[[name]],
path = here::here("2_plots/2_DE/"),
width = 13,
height = 11,
units = "cm",
dpi = 900)
}
}
saveRDS(ma, here::here("0_data/RDS_plots/ma_plots.rds"))
# display
ma[[1]]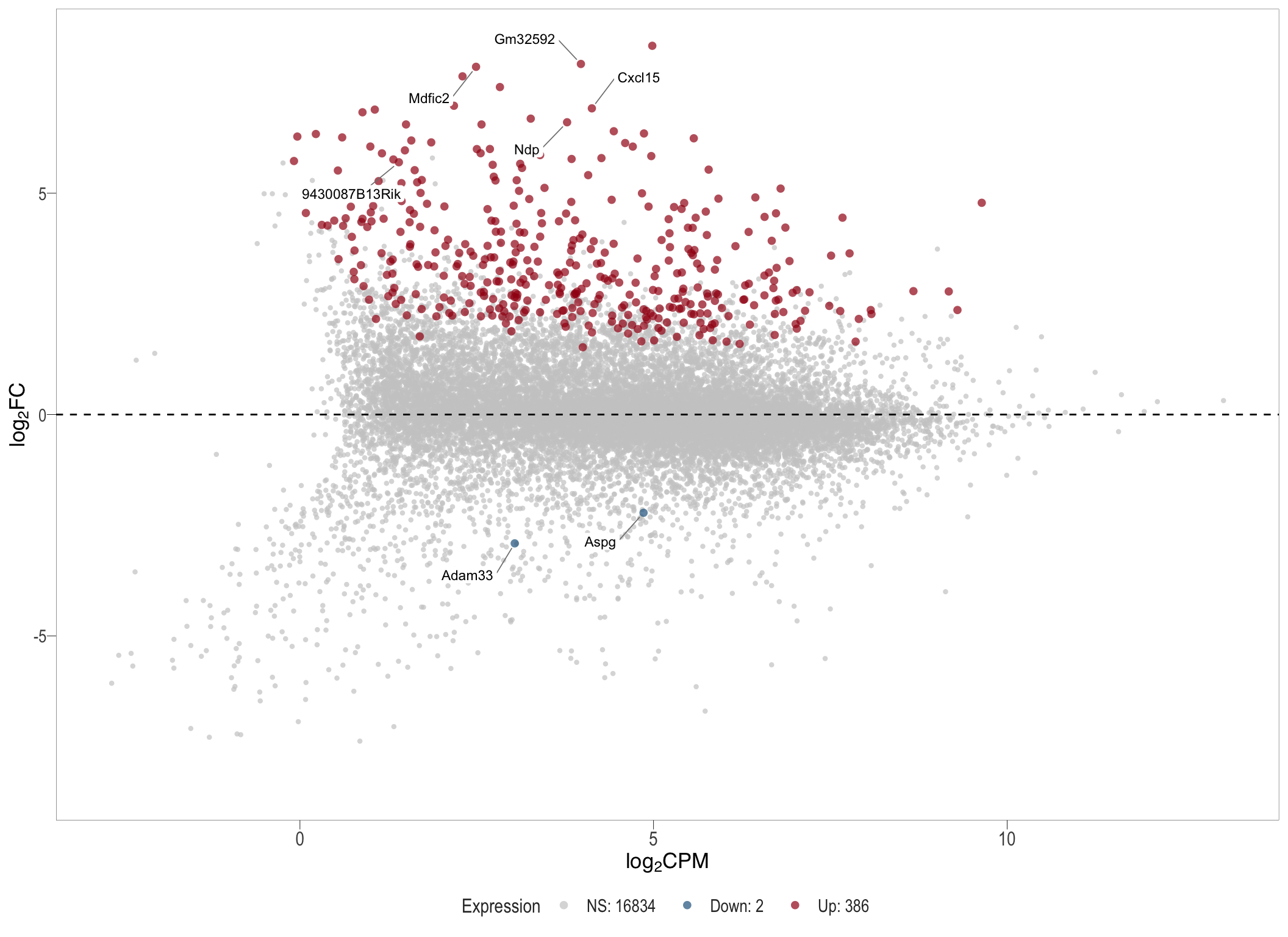
Volcano plot
library(ggrastr)
vol <- list()
for (name in comp) {
top <- 5
top_limma <- bind_rows(
lm_all[[name]] %>%
dplyr::filter(expression == "up") %>%
arrange(adj.P.Val, desc(abs(logFC))) %>%
head(top),
lm_all[[name]] %>%
dplyr::filter(expression == "down") %>%
arrange(adj.P.Val, desc(abs(logFC))) %>%
head(top)
)
invisible(top_limma %>% as.data.frame())
max <- max(lm_all[[name]]$logFC)
vol[[name]] <- lm_all[[name]] %>%
ggplot(aes(x = logFC,y = -log(adj.P.Val, 10))) +
rasterise(geom_point(aes(colour = expression, size = expression), show.legend = T, alpha =0.7, stroke =0 ), dpi = 600) +
geom_label_repel(data = top_limma,
# lm_all[[name]] %>% dplyr::filter(gene_name %in% c("Itprid1", "Scart1", "Blk", "Cd3e", "Trgc1", "Il7r","Nr4a3","Coch")),
mapping = aes(label = gene_name),
size = 5,
label.padding = 0.15,
label.size = 0,
label.r = 0.15,
box.padding = 0.9,
point.padding = 0.5,
segment.size = 0.3,
segment.color = "grey50") +
labs(x = expression("log"[2] * "FC"),
y = expression("-log"[10] * "FDR"),
colour = "Expression") +
scale_x_continuous(limits = c(-max,max),expand = expansion(mult = c(0.01, 0.01))) +
scale_size_manual(values = c(1.5,2.4,2.4), guide = "none") +
scale_fill_manual(values = expressionCol) +
scale_color_manual(labels = c(paste0("NS: ", sum(lm_all[[name]]$expression == "insig"), " "),
paste0("Down: ", sum(lm_all[[name]]$expression == "down"), " "),
paste0("Up: ", sum(lm_all[[name]]$expression == "up"), " ")),
values = expressionCol_dark) +
bossTheme( legend = "bottom") +
guides(colour = guide_legend(override.aes = list(size = 2.4)))
if (savePlots == T) {
ggsave(paste0("vol_", name, ".png"),
plot = vol[[name]],
path = here::here("2_plots/2_DE/"),
width = 11,
height = 13,
units = "cm",
dpi = 900)
}
}
saveRDS(vol, here::here("0_data/RDS_plots/vol_plots.rds"))
# display
vol[[1]]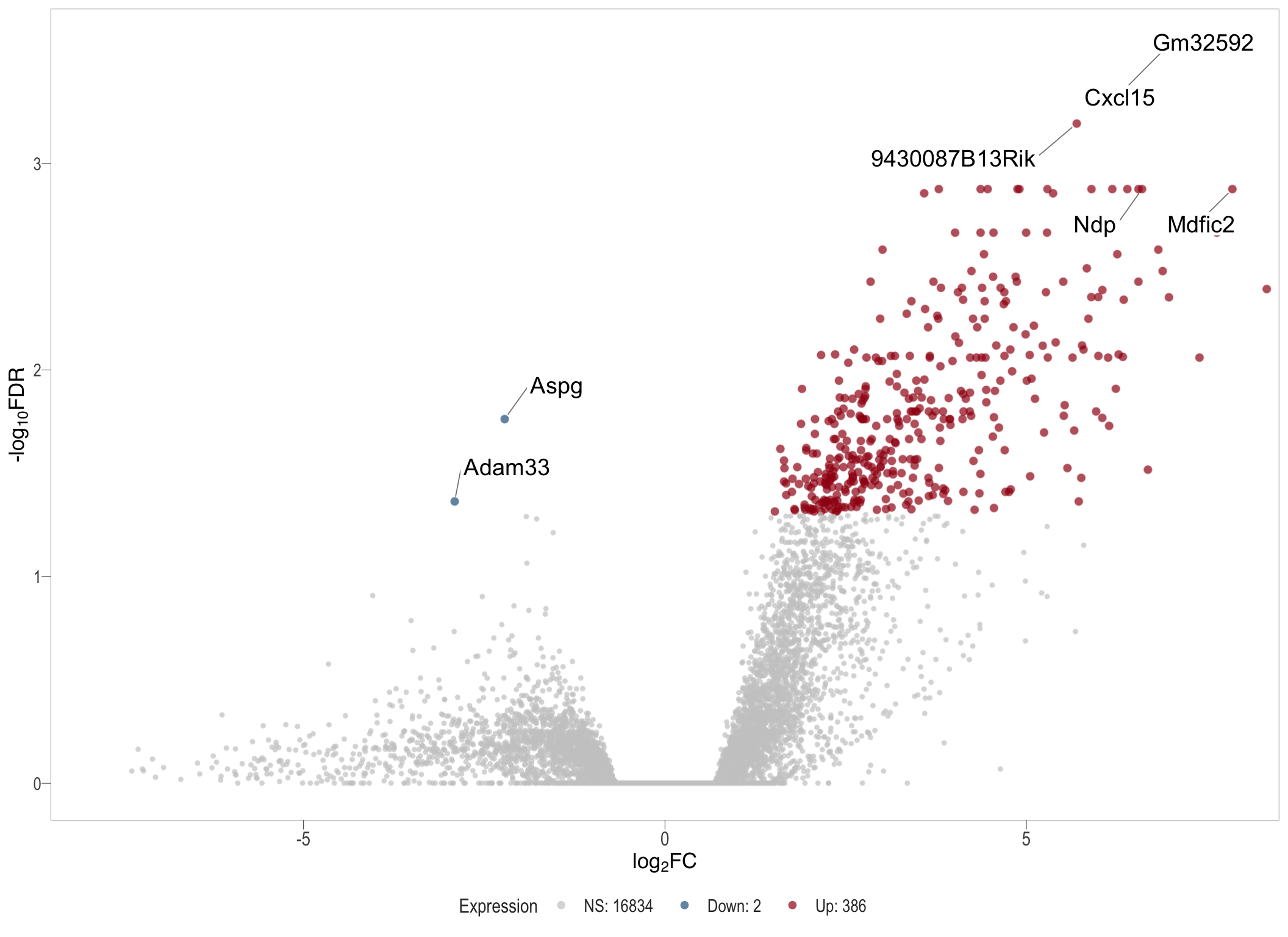
ggsave(filename = "intVsvxVAS.svg", plot = vol[[2]],path = here::here("2_plots/"), width = 12, height = 12, units = "cm")Top upregulated
logCPM_up=list()
logCPM_down=list()
anno=list()
anno_colours=list()
for (i in 1:length(comp)) {
x <- comp[i] %>% as.character()
# initialise/reinitialise the count matrix
logCPM <- cpm(dge, prior.count = 3, log = TRUE)
rownames(logCPM) <- dge$genes$gene_name
colnames(logCPM) <- dge$samples$pretty
# initialise/reinitialise the annotations used for heatmap legend
anno[[x]] <- as.factor(dge$samples$group) %>% as.data.frame()
colnames(anno[[x]]) <- "Groups"
if (i == 1) { # for the first comparison, extract just intact and control
logCPM <- subset(logCPM ,select = 1:7)
anno[[x]] <- dplyr::slice(anno[[x]], 1:7)
rownames(anno[[x]]) <- colnames(logCPM) }
if (i == 2) { # for the second comapsion, extract intact and SVX_VAS
logCPM <- subset(logCPM, select = c(4:7,12:15))
anno[[x]] <- dplyr::slice(anno[[x]], c(4:7,12:15))
rownames(anno[[x]]) <- colnames(logCPM) }
if (i == 3) { # for the second comapsion, extract SVX and SVX_VAS
logCPM <- subset(logCPM, select = 8:15)
anno[[x]] <- dplyr::slice(anno[[x]], 8:15)
rownames(anno[[x]]) <- colnames(logCPM) }
if (i == 4) { # for the second comapsion, extract VAS and SVX_VAS
logCPM <- subset(logCPM, select = c(16:19, 12:15))
anno[[x]] <- dplyr::slice(anno[[x]], c(16:19, 12:15))
rownames(anno[[x]]) <- colnames(logCPM) }
if (i == 5) { # for the first comparison, extract just intact and control
logCPM <- subset(logCPM ,select = c(12:15,1:3))
anno[[x]] <- dplyr::slice(anno[[x]], c(12:15,1:3))
rownames(anno[[x]]) <- colnames(logCPM) }
if (i == 6) { # for the first comparison, extract just intact and control
logCPM <- subset(logCPM ,select = c(4:7,16:19))
anno[[x]] <- dplyr::slice(anno[[x]], c(4:7,16:19))
rownames(anno[[x]]) <- colnames(logCPM) }
# filtering top unregulated genes then filter the logCPM values of those genes.
upReg <- lm_sig[[x]] %>%
dplyr::filter(logFC > 0) %>%
arrange(sort(adj.P.Val, decreasing = F))
upReg <- upReg[1:20,]
logCPM_up[[x]] <- logCPM[upReg$gene_name,] %>% as.data.frame()
# filtering top unregulated genes then filter the logCPM values of those genes.
downReg <- lm_sig[[x]] %>%
dplyr::filter(logFC < 0) %>%
arrange(sort(adj.P.Val, decreasing = F))
if (nrow(downReg) >= 20) {max <- 20} else {max <- nrow(downReg)}
downReg <- downReg[1:max,] %>% na.omit()
logCPM_down[[x]] <- logCPM[downReg$gene_name,] %>% na.omit %>% as.data.frame()
}library(ggplotify)
heat_up=list()
custom_heatUp <- function(direction, ...){
if (direction == "up") {
mat <- logCPM_up[[x]]
} else {
mat <- logCPM_down[[x]]
}
ComplexHeatmap::pheatmap(
mat = mat,
color = colorRampPalette(rev(c("#FB8072","#FDB462","#ffffd5","#8DD3C7","#80B1D3")))(100),
# cellheight = 20,
cellwidth = 30,
scale = "row",
border_color = "white",
treeheight_row = 40,
treeheight_col = 30,
show_colnames = T,
clustering_distance_rows = "euclidean",
main = paste0("Top ", nrow(mat), " significant ", direction,"regulated genes\n"),
legend = F,
# heatmap_legend_param = list(title = "Z-score",
# legend_direction = "vertical",
# legend_width = unit(5, "cm")),
# annotation = T,
annotation_legend = T,
annotation_col = anno[[x]],
annotation_colors = list("Groups" = groupColour),
annotation_names_col = F,
fontfamily = "Arial Narrow",
fontsize = 14,
fontsize_col = 14,
fontsize_number = 14,
fontsize_row = 14,
labels_row = as.expression(lapply(rownames(mat), function(a) bquote(italic(.(a))))),
...) %>% as.ggplot()
}
for (i in 1:length(comp)) {
x <- comp[i] %>% as.character()
if (i == 1 || 5) {
heat_up[[x]] <- custom_heatUp(direction = "up",
cluster_cols = T,
cutree_cols = 2,
cutree_rows = 4
)
} else {
heat_up[[x]] <- custom_heatUp(direction = "up",
cluster_cols = T,
cutree_cols = 2,
cutree_rows = 4,
# gaps_col = c(4)
)
}
ggsave(paste0("heat_up_", comp[i], ".svg"),
plot = heat_up[[x]],
path = here::here("2_plots/2_DE/"),
width = 166,
height = 250,
units = "mm"
)
}
heat_up[[1]] 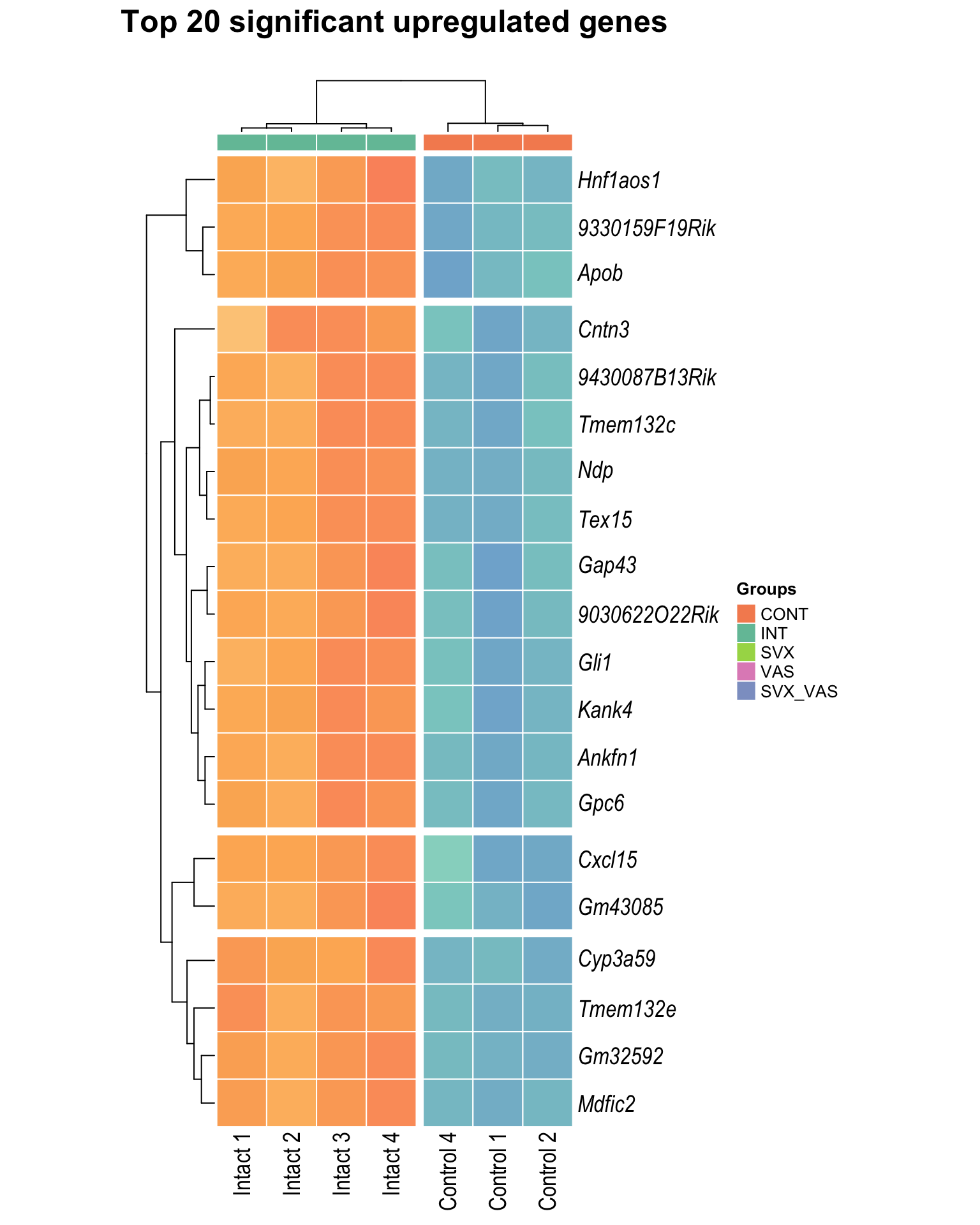
Top downregulated
heat_down=list()
for (i in 1:length(comp)) {
x <- comp[i] %>% as.character()
if (i == 1 || 5) {
heat_down[[x]] <- custom_heatUp(direction = "down",
cluster_cols = T,
cutree_cols = 2,
cutree_rows = 2
) %>% as.ggplot()
} else {
heat_down[[x]] <- custom_heatUp(direction = "down",
cluster_cols = T,
cutree_cols = 2,
cutree_rows = 4,
# gaps_col = c(4)
) %>% as.ggplot()
}
ggsave(paste0("heat_down_", comp[i], ".svg"),
plot = heat_down[[x]],
path = here::here("2_plots/2_DE/"),
width = 166,
height = 250,
units = "mm"
)
}
heat_down[[1]] 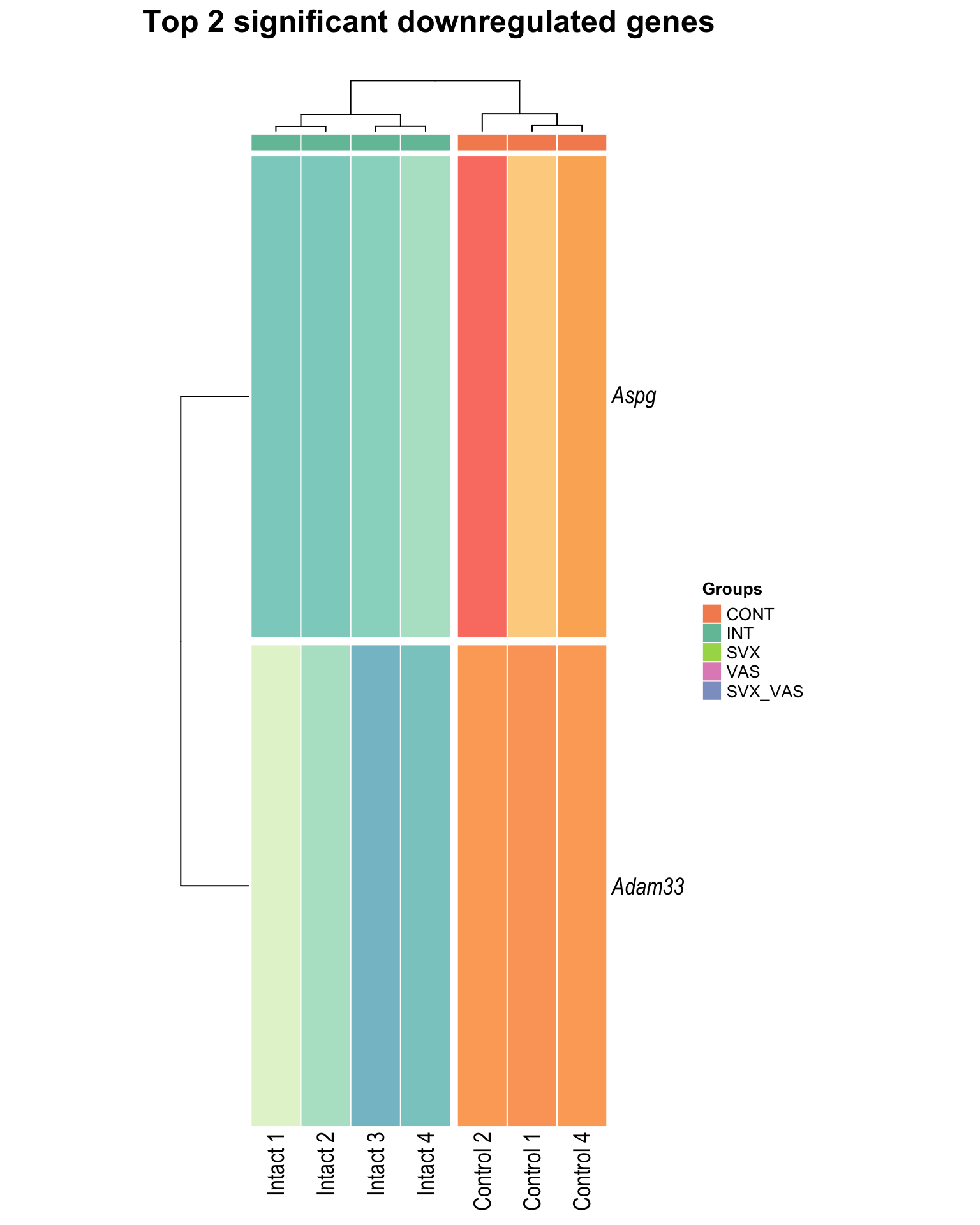
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
| ae93fcc | tranmanhha135 | 2024-02-02 |
| 2c24612 | tranmanhha135 | 2022-10-13 |
| 11a5cf4 | tranmanhha135 | 2022-10-03 |
| 1101367 | tranmanhha135 | 2022-10-02 |
| 192d010 | tranmanhha135 | 2022-09-20 |
| 959a5df | tranmanhha135 | 2022-09-07 |
| 32454d5 | Ha Manh Tran | 2022-01-01 |
| 3b268fc | Ha Tran | 2021-12-09 |
| f4ba25b | Ha Manh Tran | 2021-11-23 |
## this function is basically creating chunks within chunks, and then
## I use results='asis' so that the html image code is rendered
library(knitr)
kexpand <- function(wd, ht, cap) {
cat(knit(text = knit_expand(text =
sprintf("```{r %s, fig.width=%s, fig.height=%s}\n.pl\n```", cap,wd, ht)
)))}
# Loop through each FC value
types <- c("P-val histogram", "MA plot", "Volcano plot", "Top upregulated", "Top downregulated")
for (i in 2:length(comp)) {
cat(paste0("## ",comp[i],"{.tabset .tabset-pills} \n\n"))
cat(paste0("### ",types[[1]]," \n"))
.pl <- lm_hist[[i]]
kexpand(wd = 10,ht = 6,cap = paste0("hist_",i))
cat("\n\n")
cat(paste0("### ",types[[2]]," \n"))
.pl <- ma[[i]]
kexpand(wd = 10,ht = 6,cap = paste0("ma_",i))
cat("\n\n")
cat(paste0("### ",types[[3]]," \n"))
.pl <- vol[[i]]
kexpand(wd = 10,ht = 6,cap = paste0("vol_",i))
cat("\n\n")
cat(paste0("### ",types[[4]]," \n"))
.pl <- heat_up[[i]]
kexpand(wd = 8,ht = 10,cap = paste0("hmap_up",i))
cat("\n\n")
cat(paste0("### ",types[[5]]," \n"))
.pl <- heat_down[[i]]
kexpand(wd = 8,ht = 10,cap = paste0("hmap_down",i))
cat("\n\n")
}INT vs SVX_VAS
P-val histogram
| | | 0% | |…………………………………………………….| 100% [hist_2]
.pl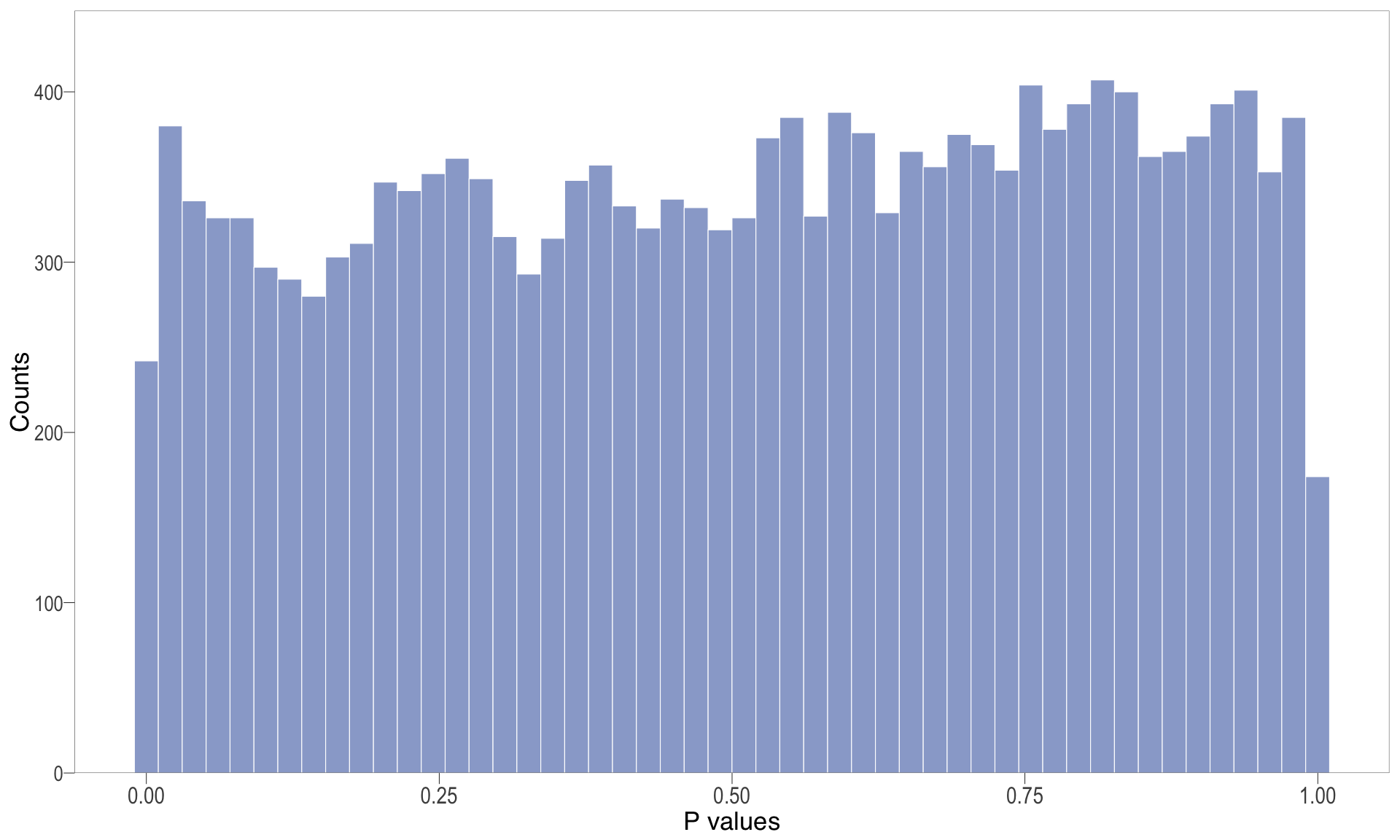
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
MA plot
| | | 0% | |………………………………………………………| 100% [ma_2]
.pl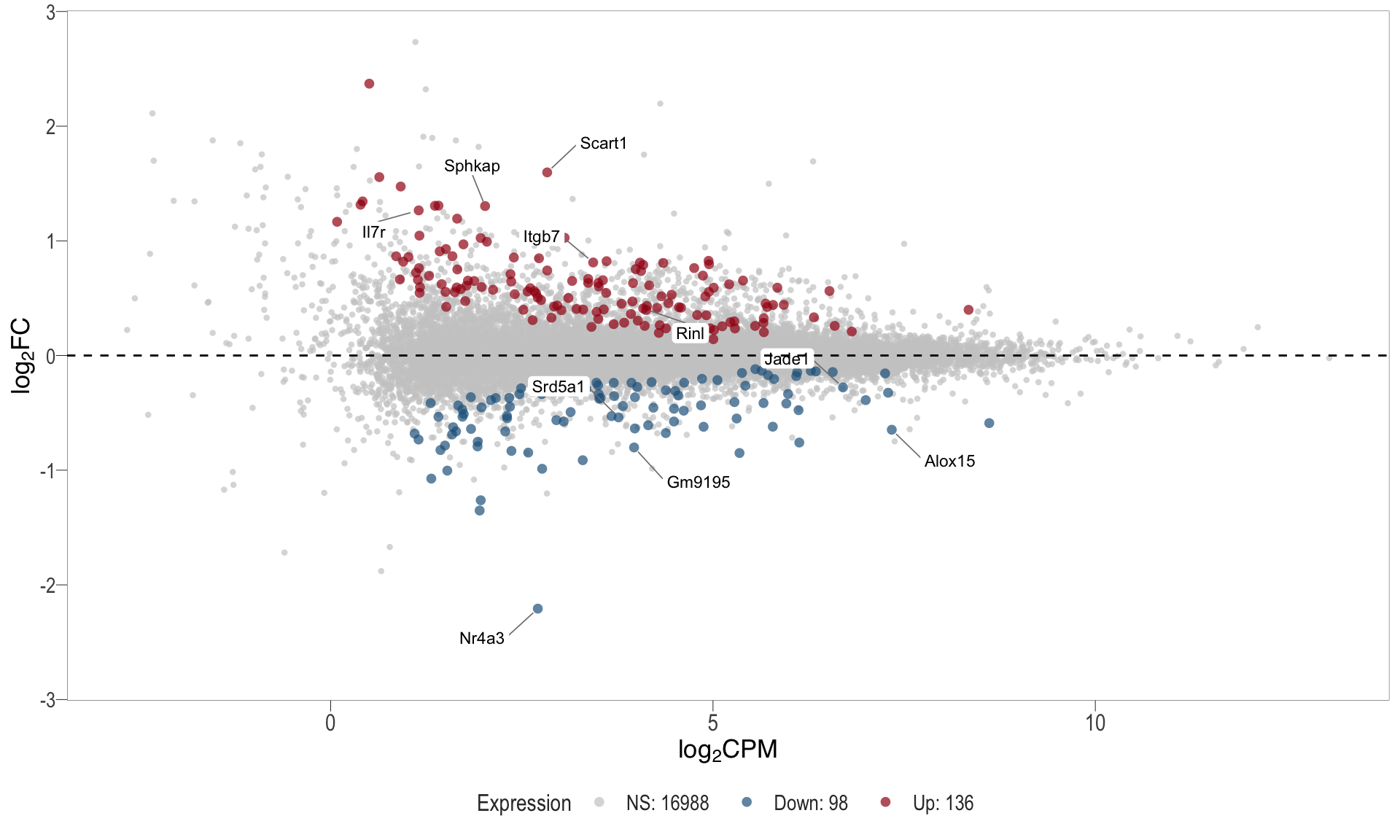
Volcano plot
| | | 0% | |……………………………………………………..| 100% [vol_2]
.pl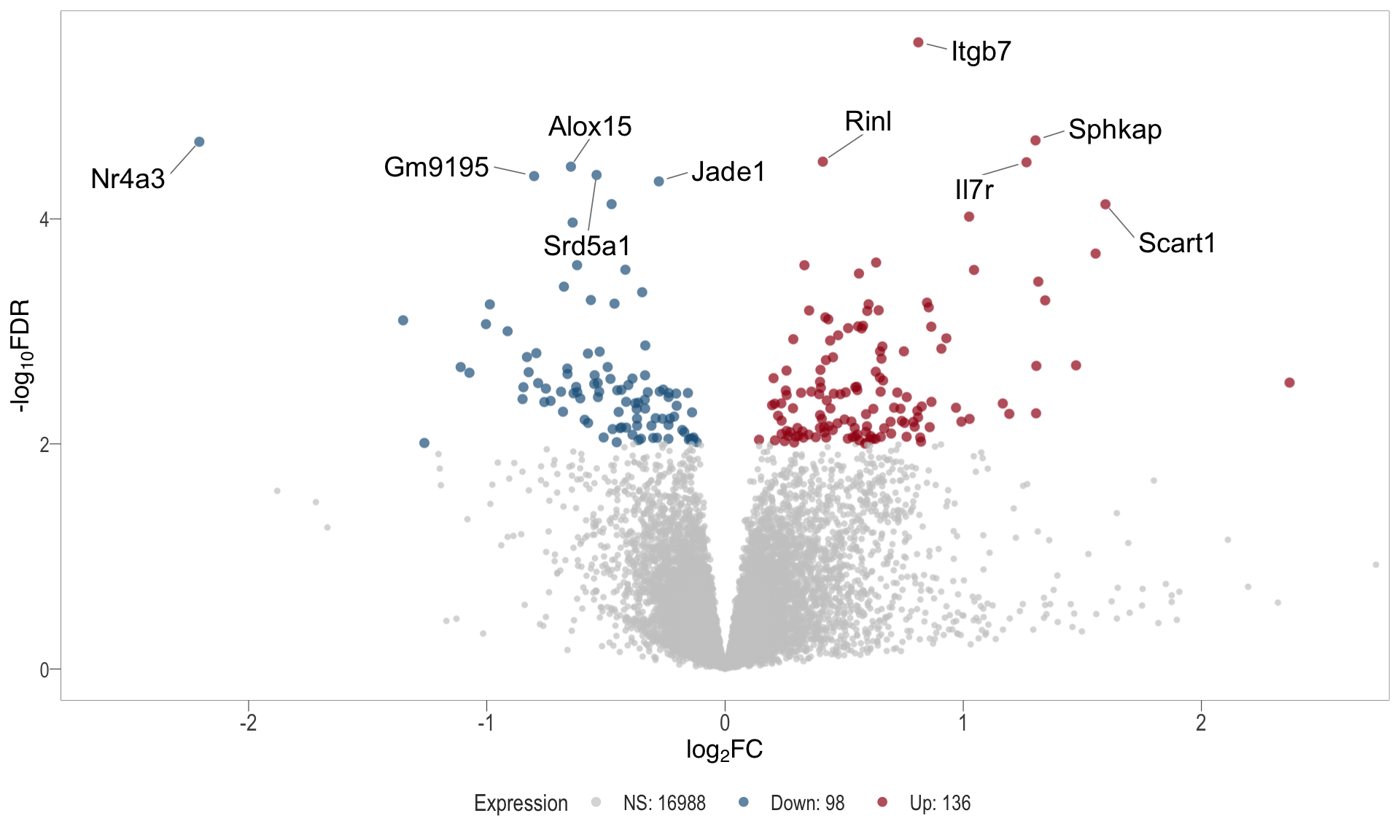
Top upregulated
| | | 0% | |…………………………………………………..| 100% [hmap_up2]
.pl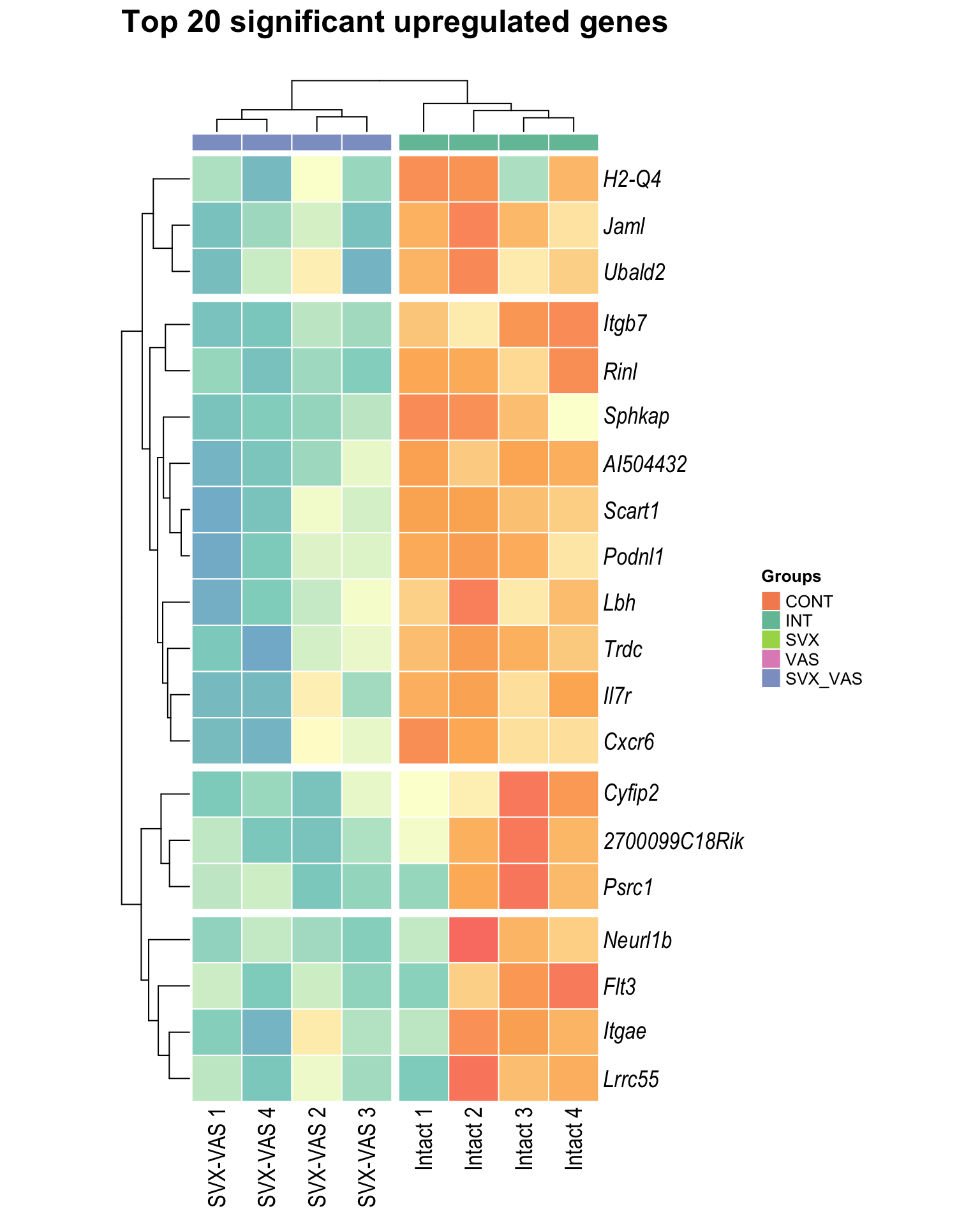
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
Top downregulated
| | | 0% | |…………………………………………………| 100% [hmap_down2]
.pl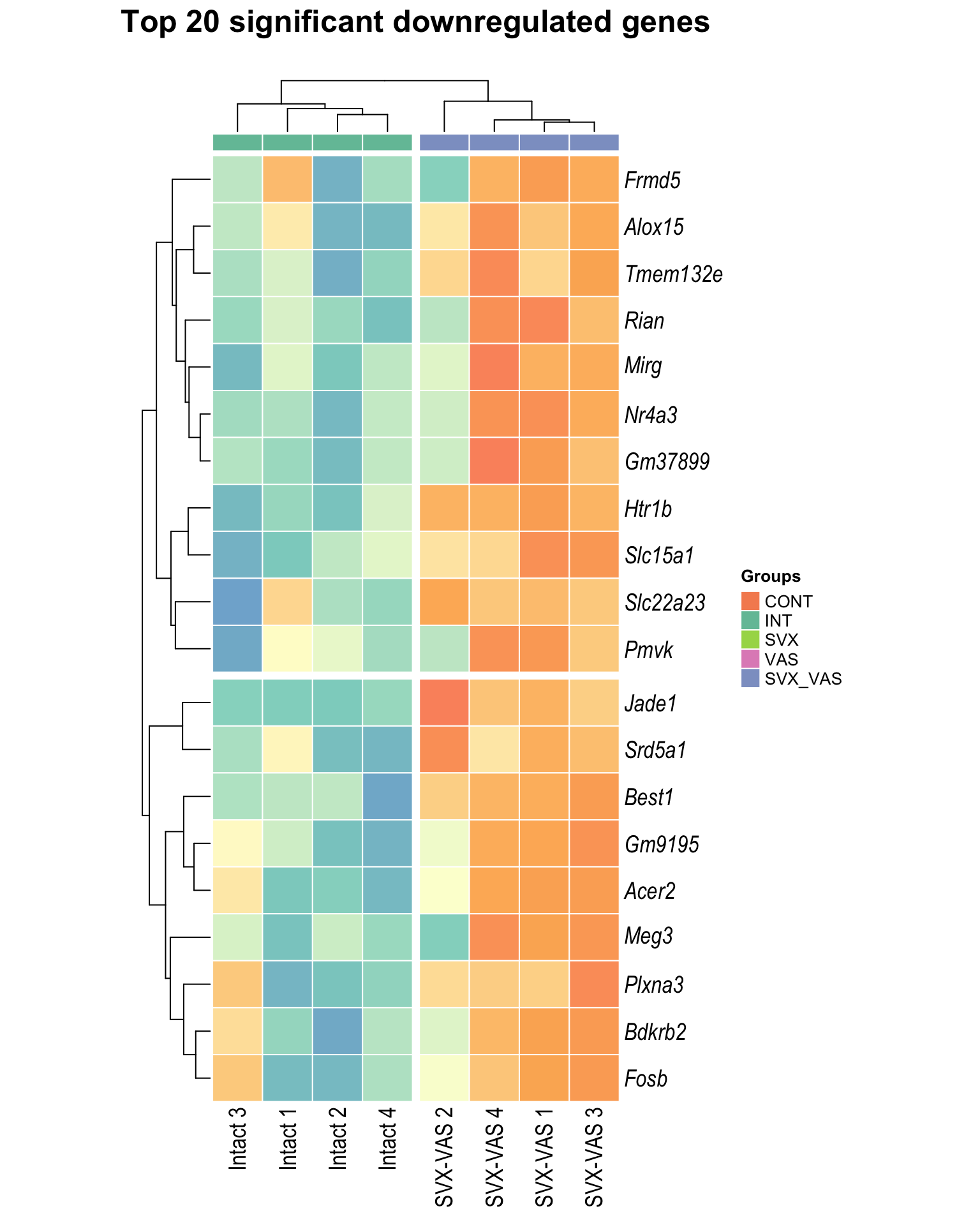
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
SVX vs SVX_VAS
P-val histogram
| | | 0% | |…………………………………………………….| 100% [hist_3]
.pl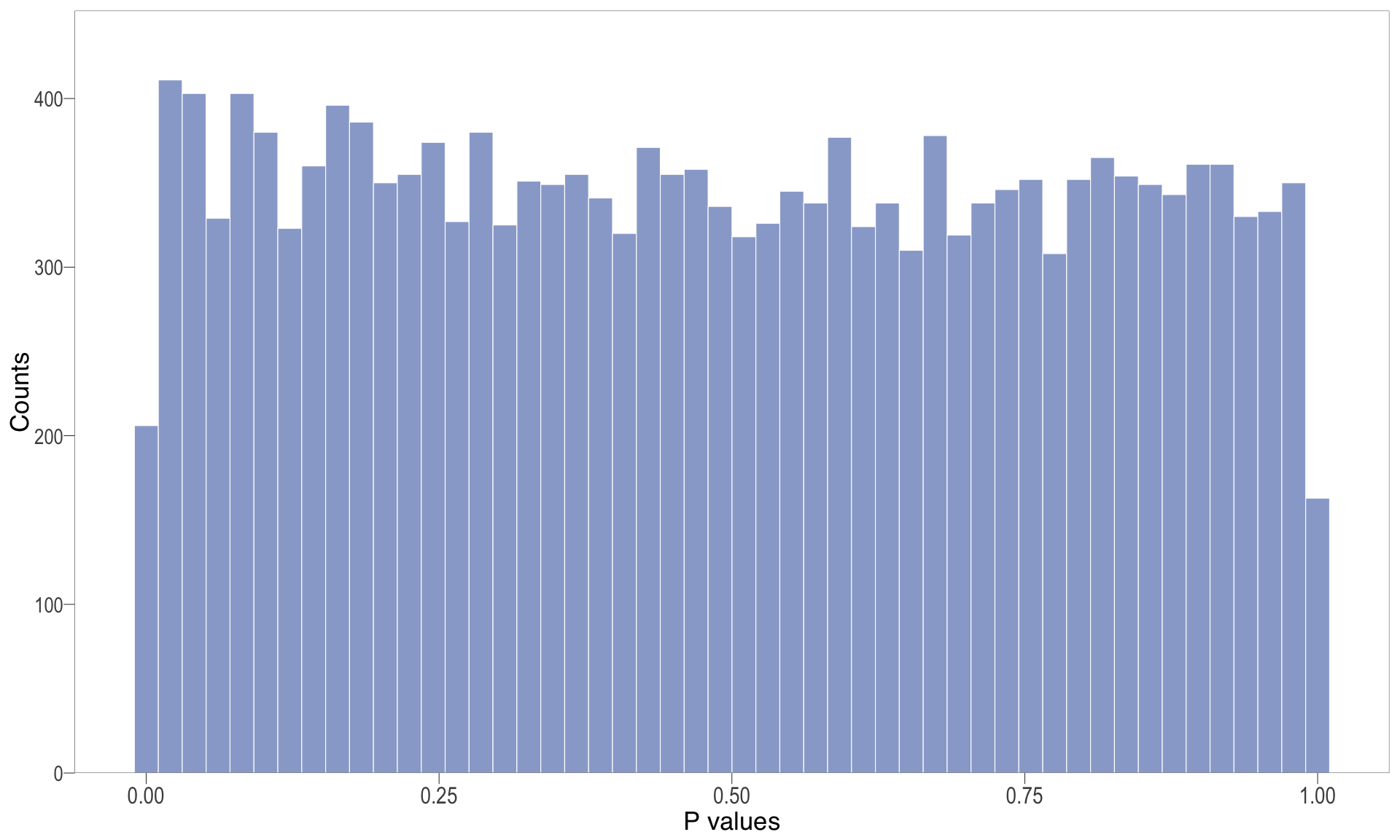
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
MA plot
| | | 0% | |………………………………………………………| 100% [ma_3]
.pl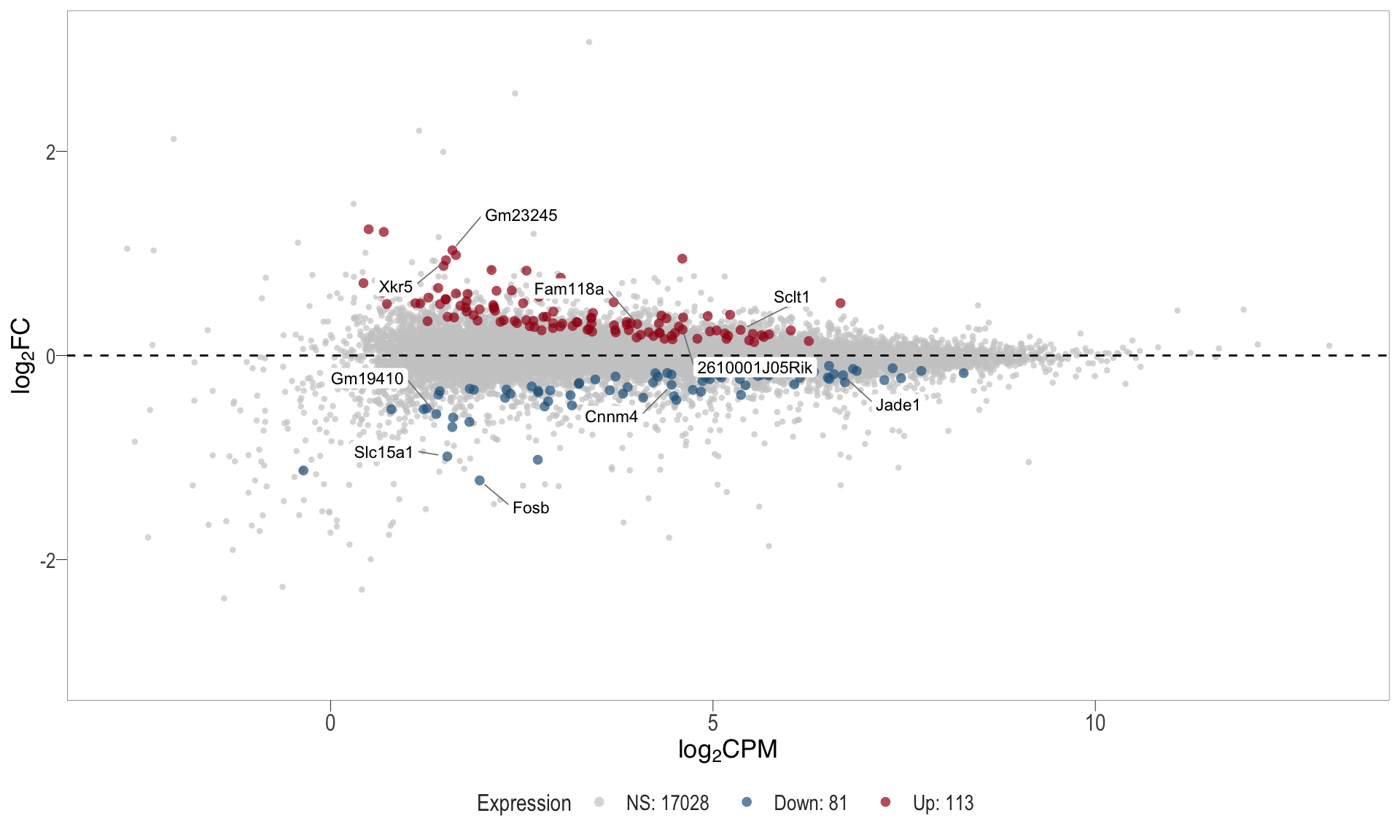
Volcano plot
| | | 0% | |……………………………………………………..| 100% [vol_3]
.pl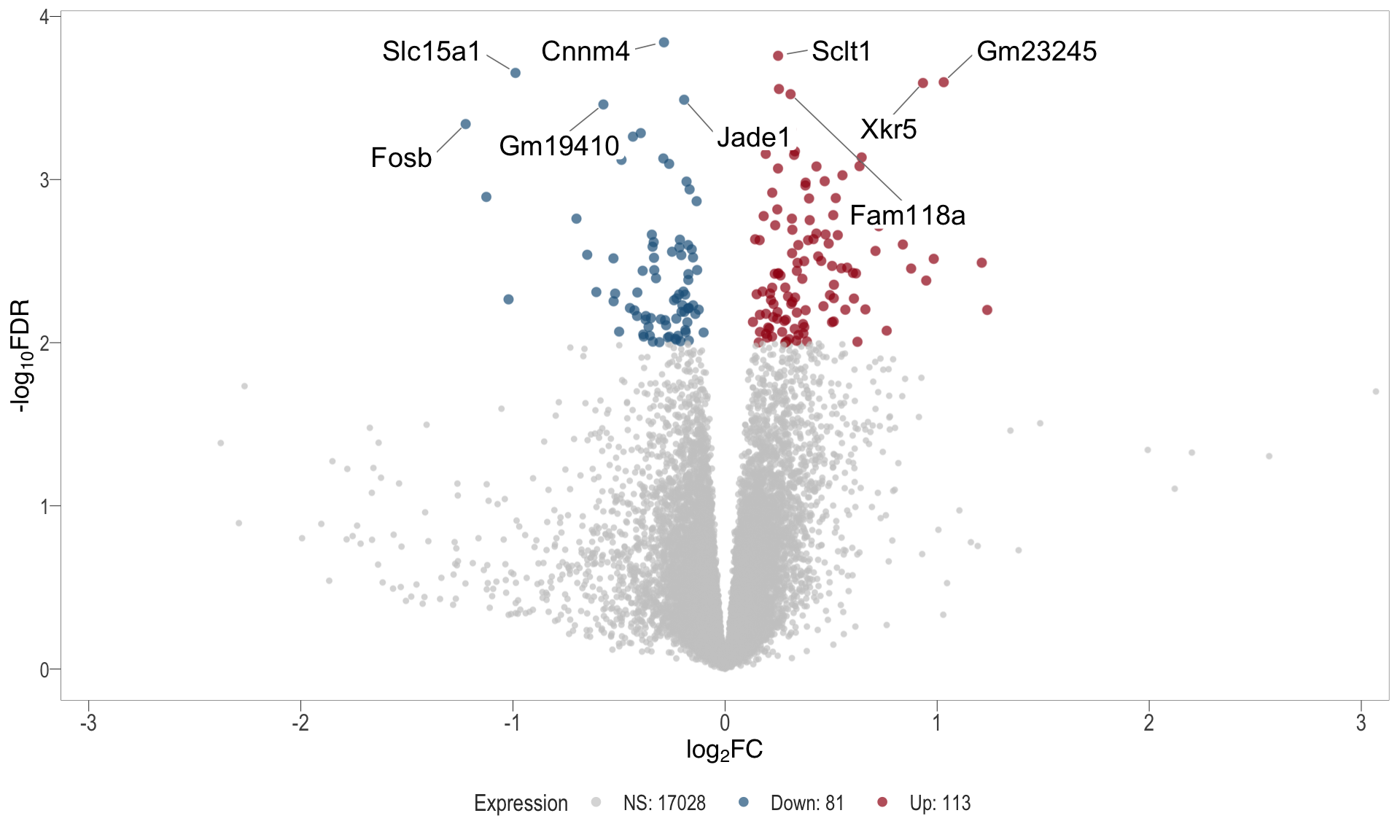
Top upregulated
| | | 0% | |…………………………………………………..| 100% [hmap_up3]
.pl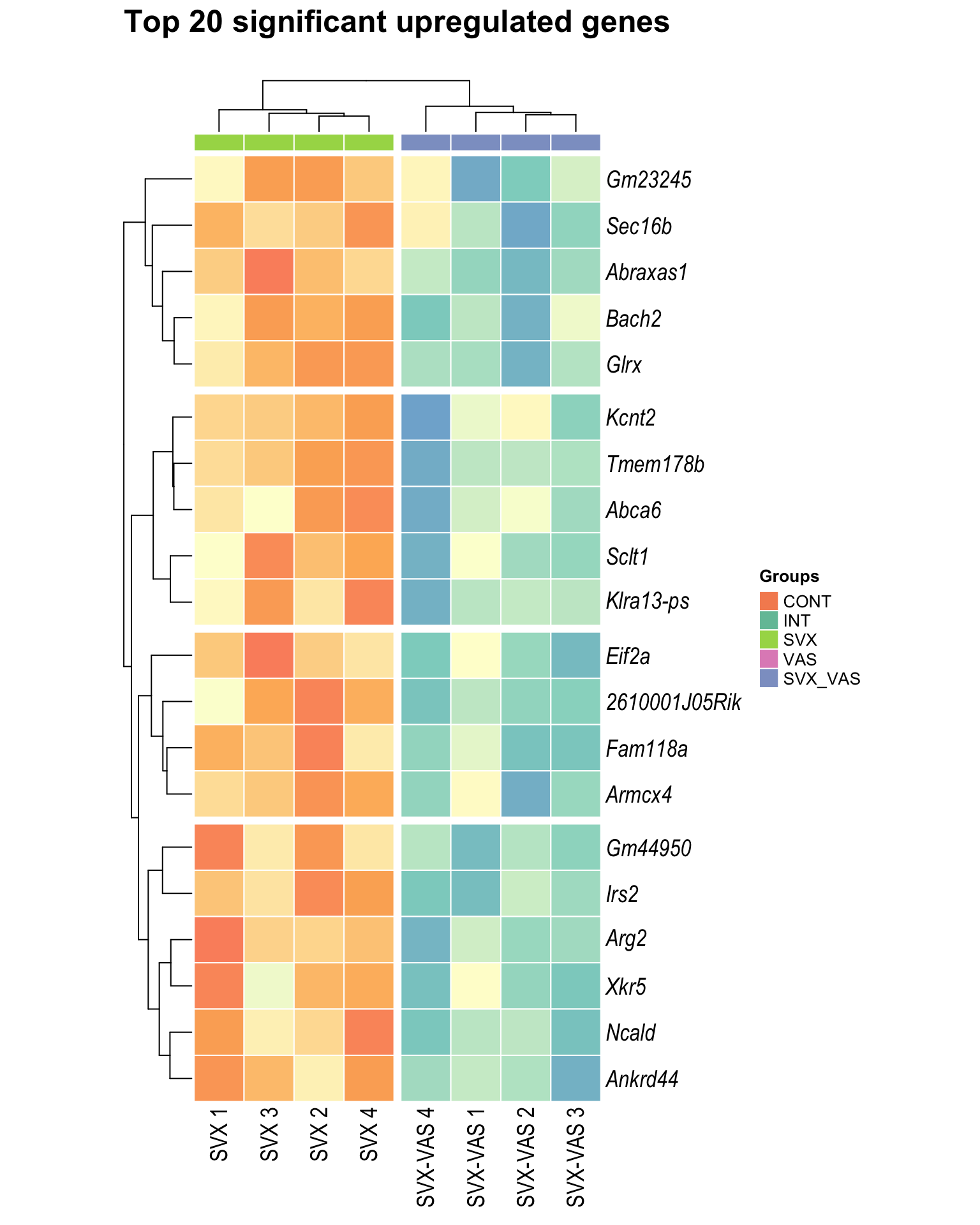
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
Top downregulated
| | | 0% | |…………………………………………………| 100% [hmap_down3]
.pl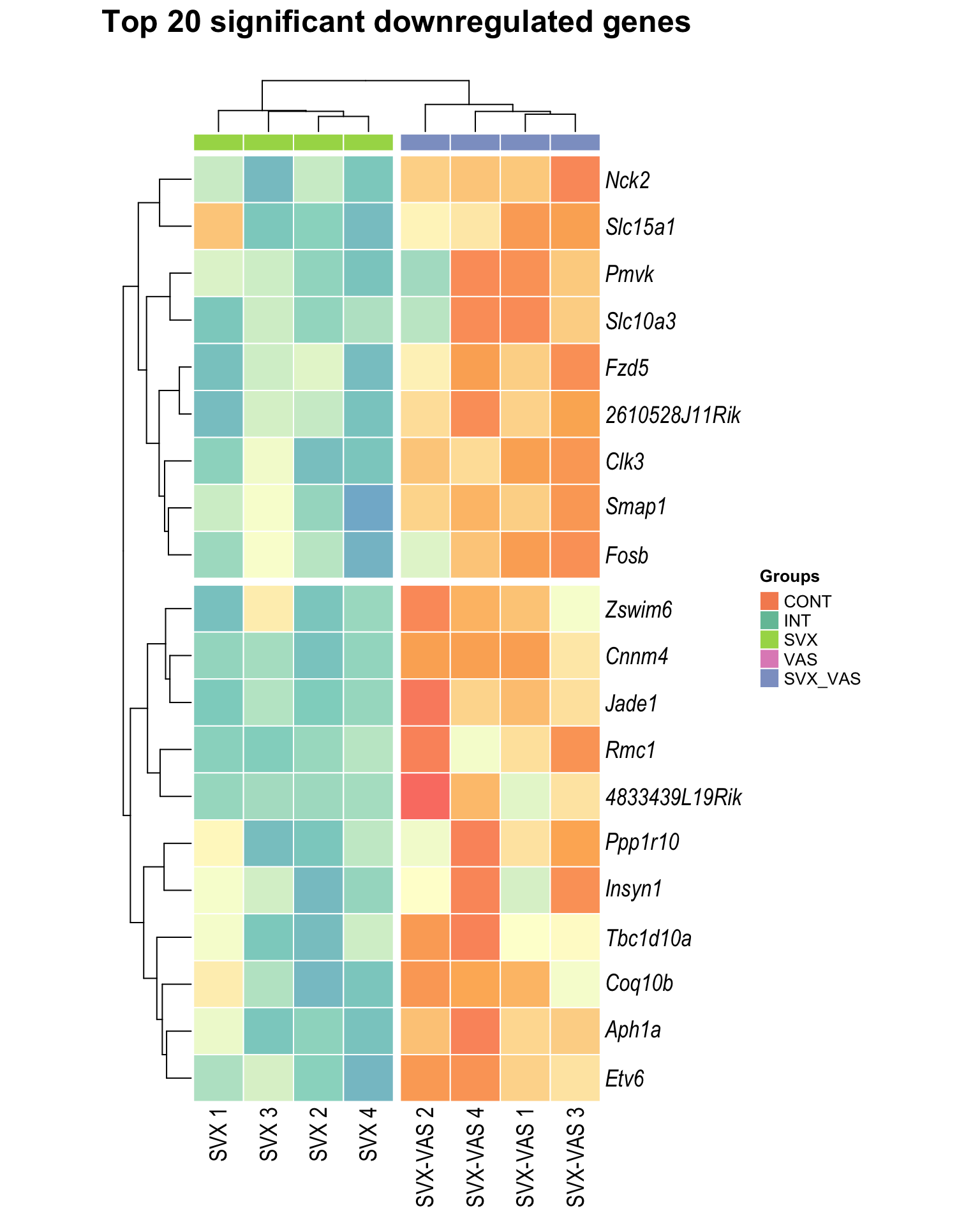
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
VAS vs SVX_VAS
P-val histogram
| | | 0% | |…………………………………………………….| 100% [hist_4]
.pl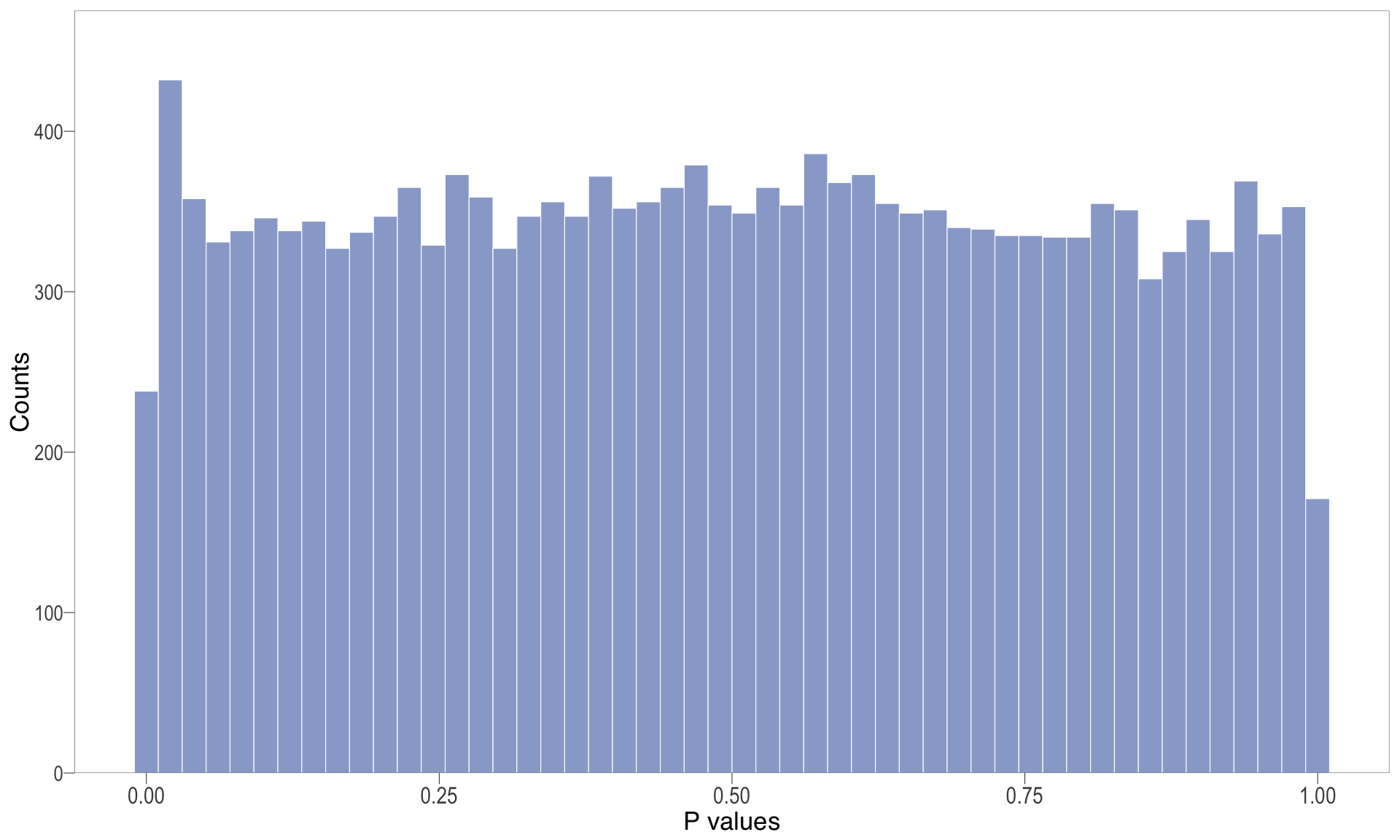
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
MA plot
| | | 0% | |………………………………………………………| 100% [ma_4]
.pl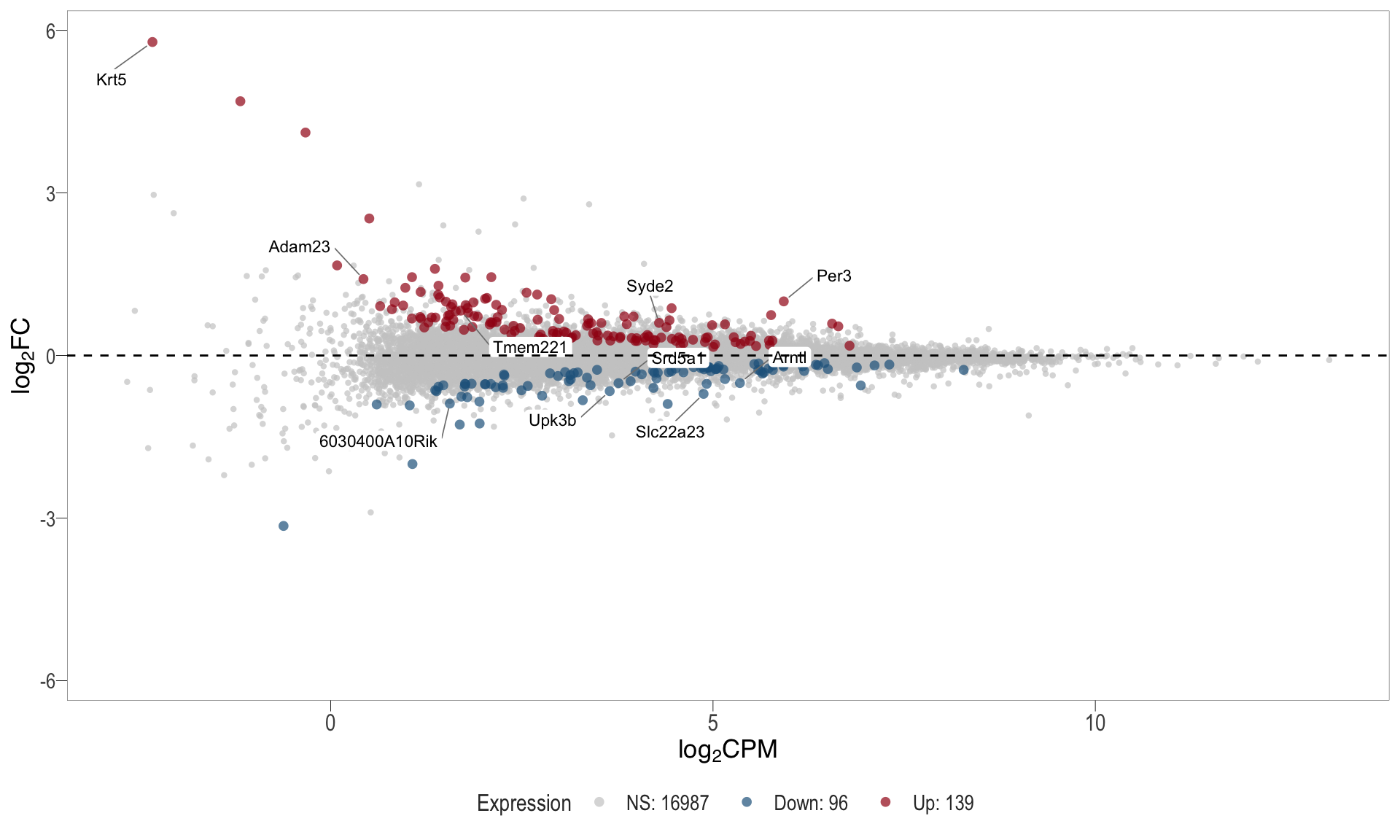
Volcano plot
| | | 0% | |……………………………………………………..| 100% [vol_4]
.pl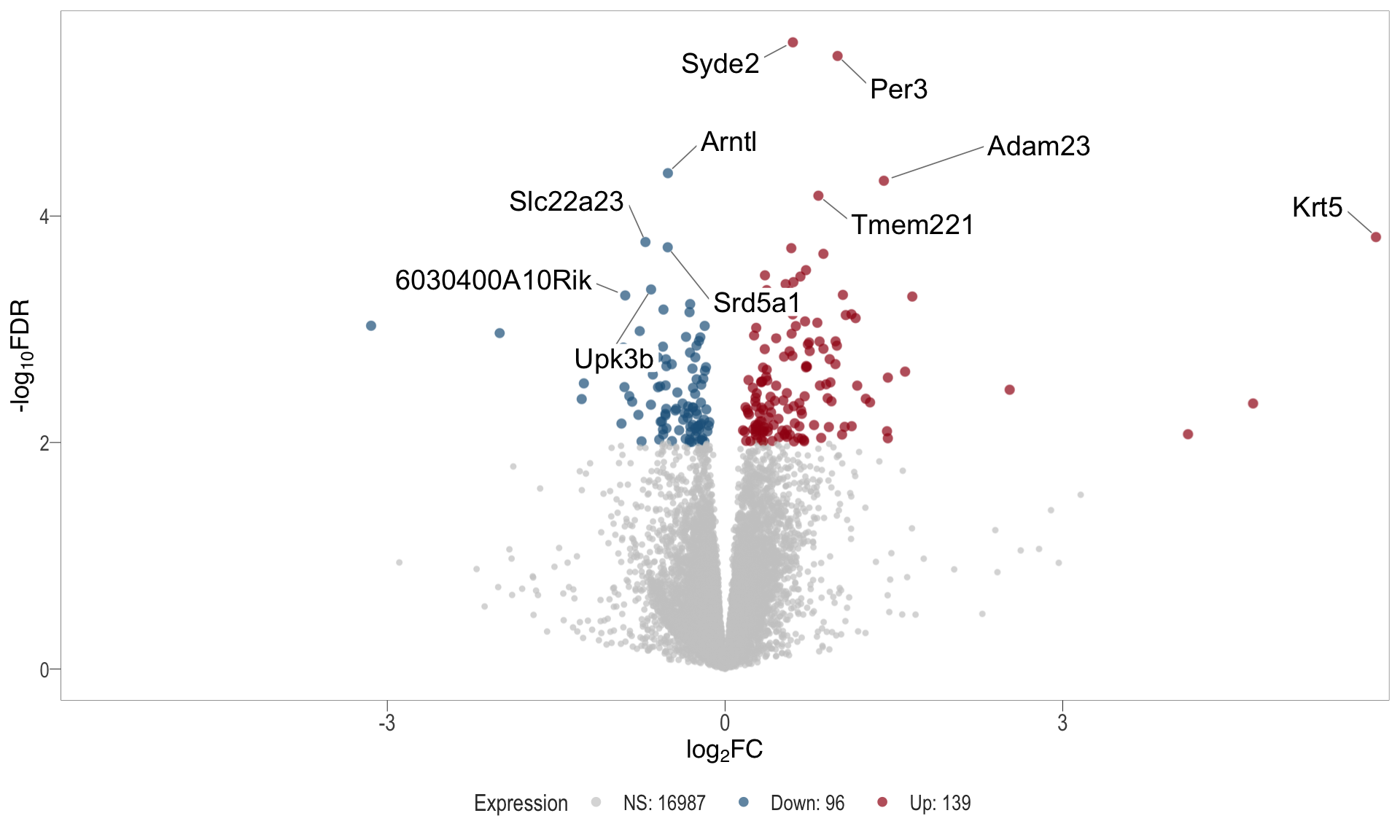
Top upregulated
| | | 0% | |…………………………………………………..| 100% [hmap_up4]
.pl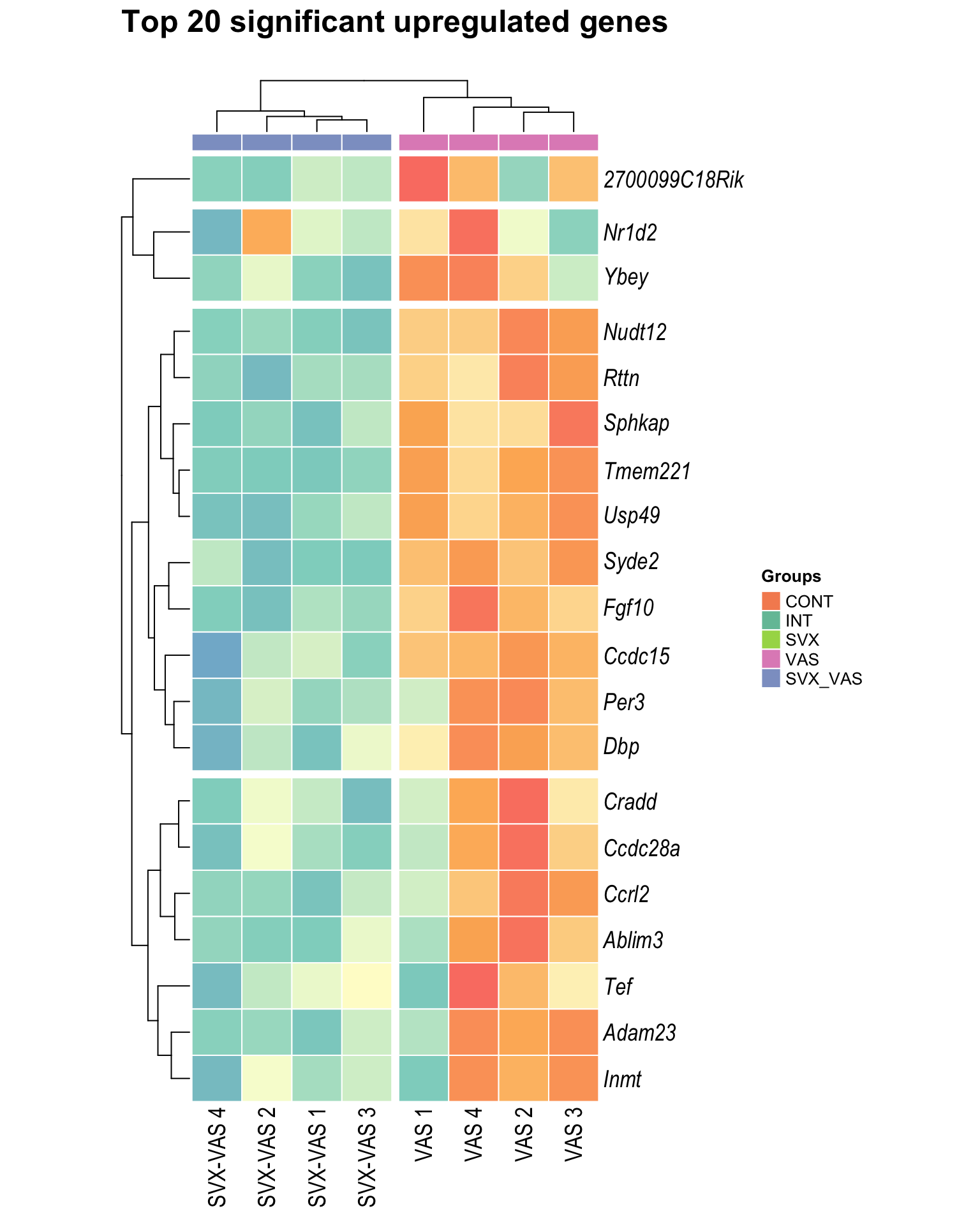
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
Top downregulated
| | | 0% | |…………………………………………………| 100% [hmap_down4]
.pl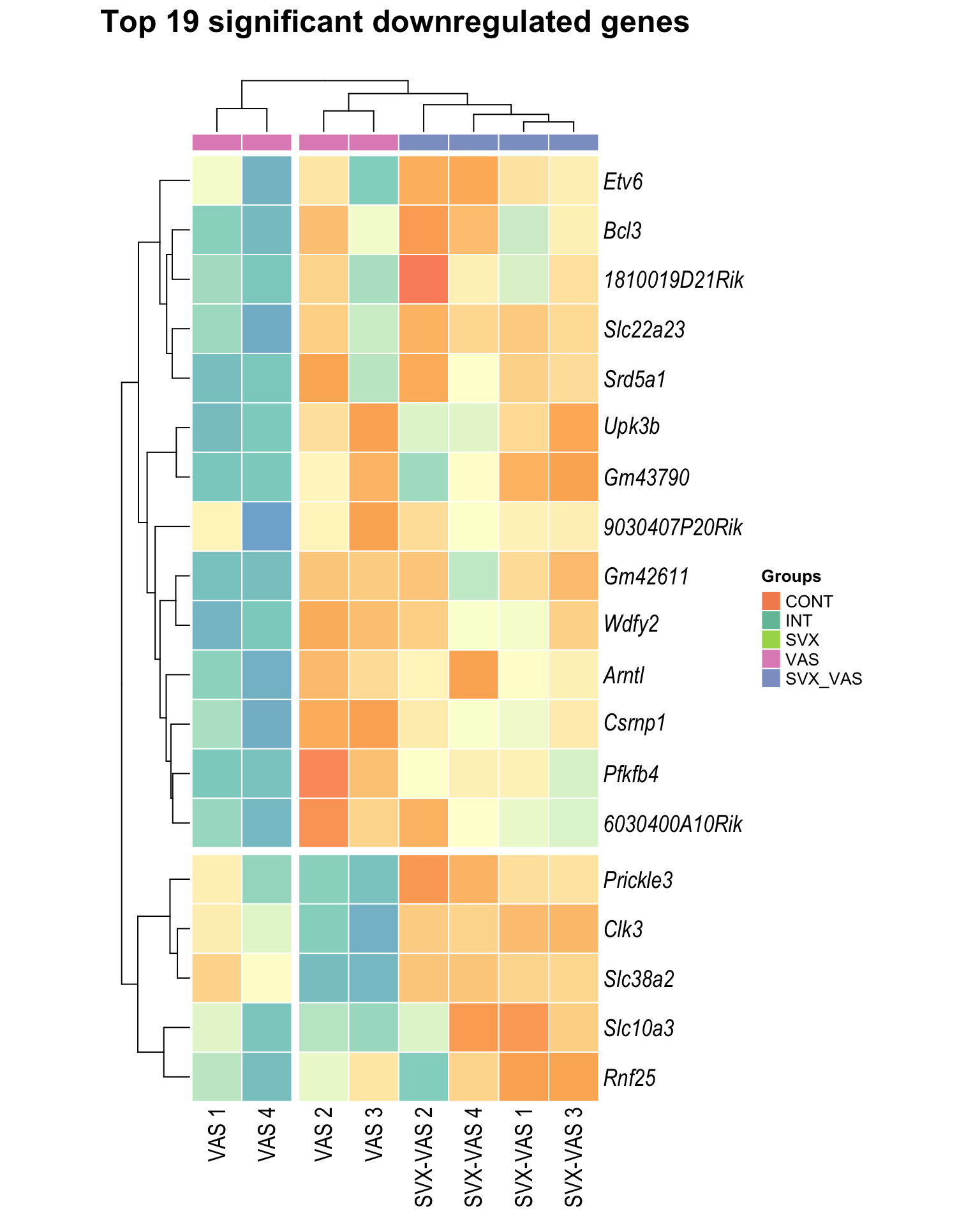
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
SVX_VAS vs CONT
P-val histogram
| | | 0% | |…………………………………………………….| 100% [hist_5]
.pl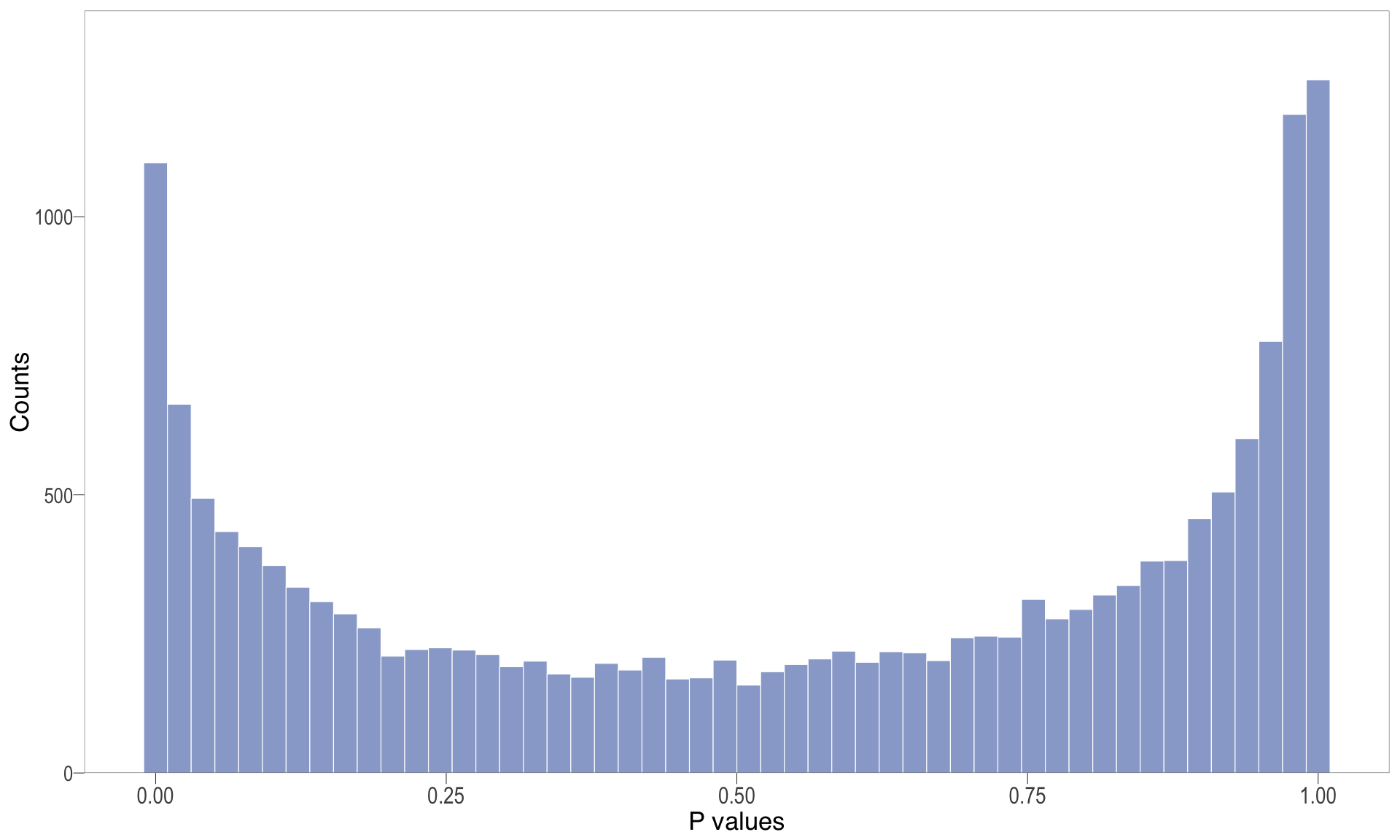
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
MA plot
| | | 0% | |………………………………………………………| 100% [ma_5]
.pl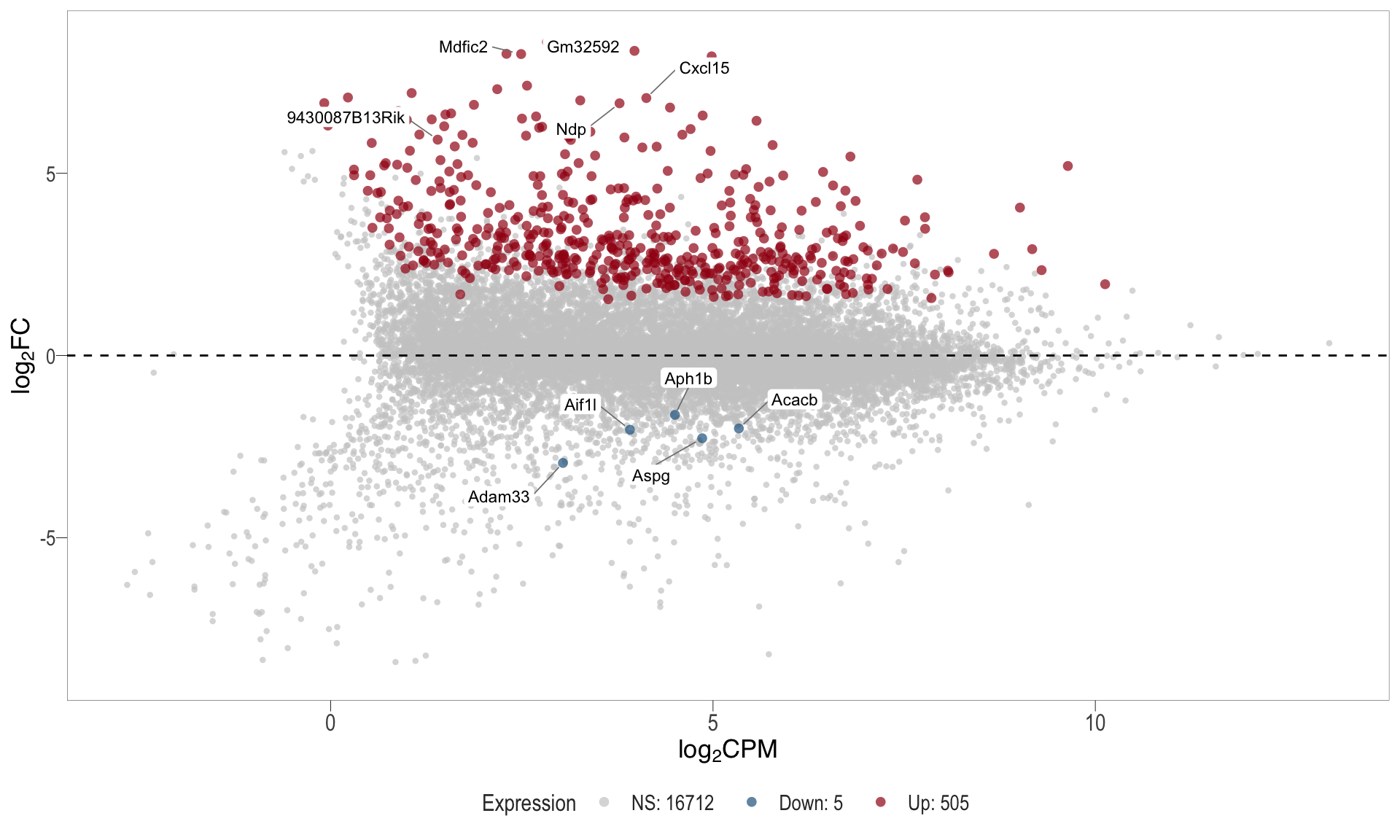
Volcano plot
| | | 0% | |……………………………………………………..| 100% [vol_5]
.pl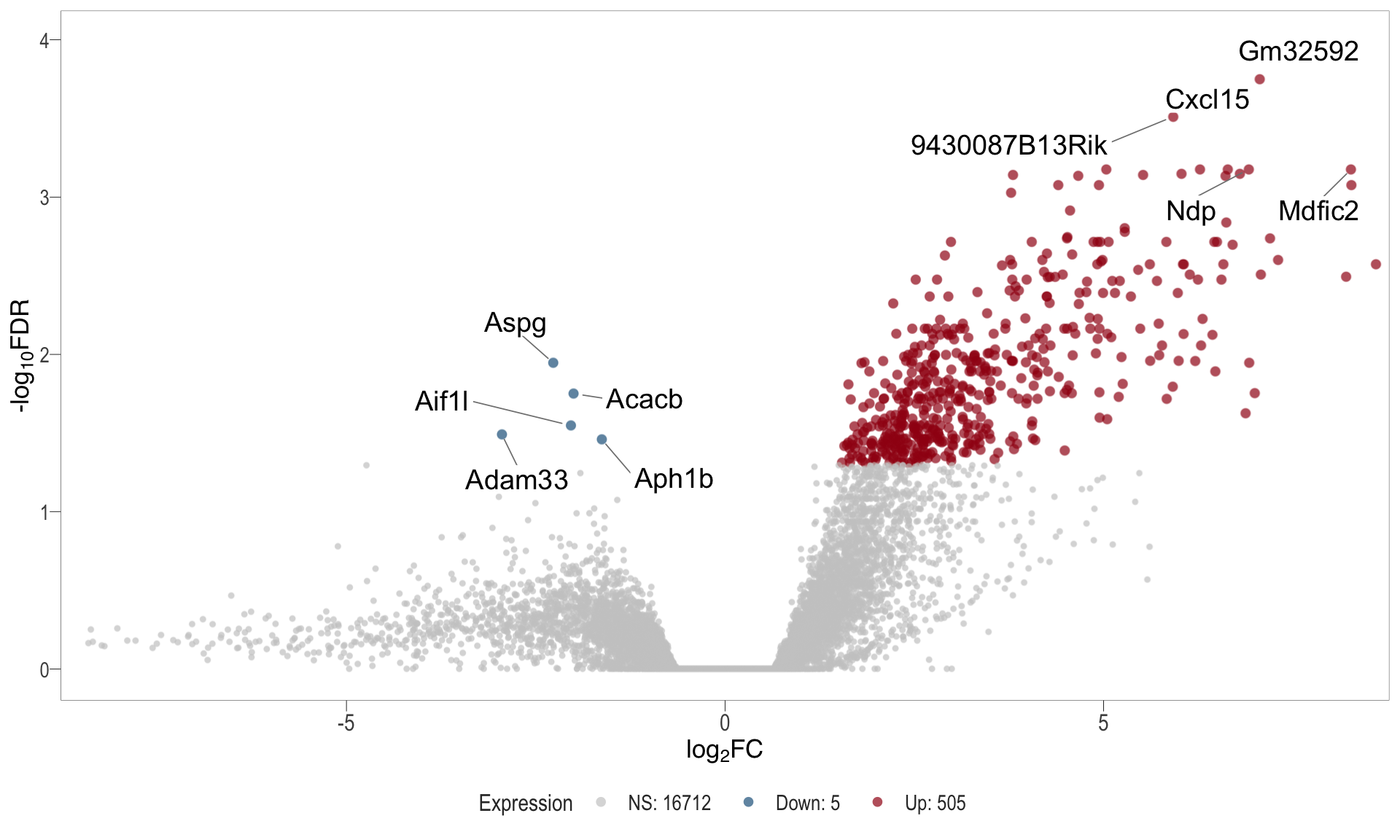
Top upregulated
| | | 0% | |…………………………………………………..| 100% [hmap_up5]
.pl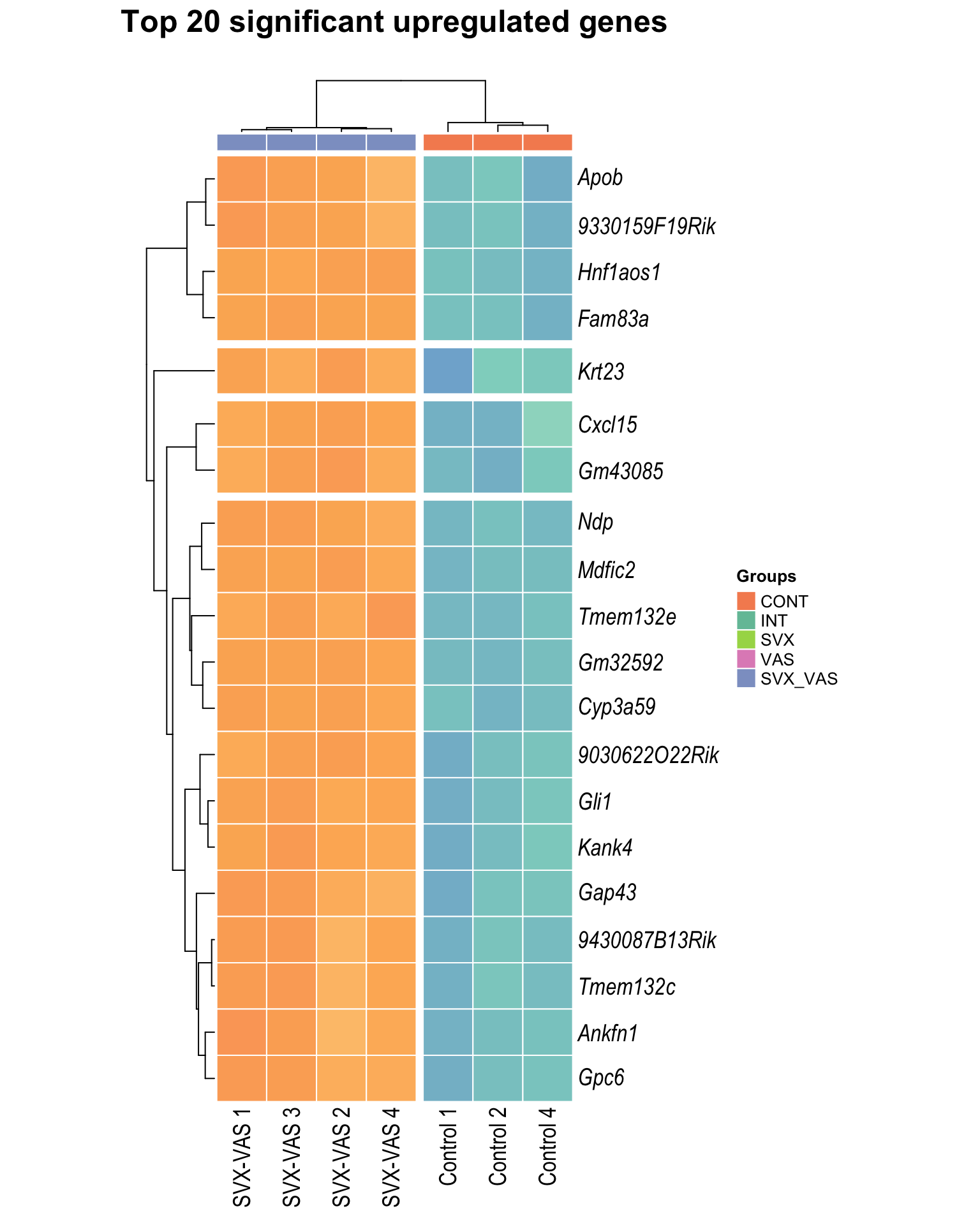
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
Top downregulated
| | | 0% | |…………………………………………………| 100% [hmap_down5]
.pl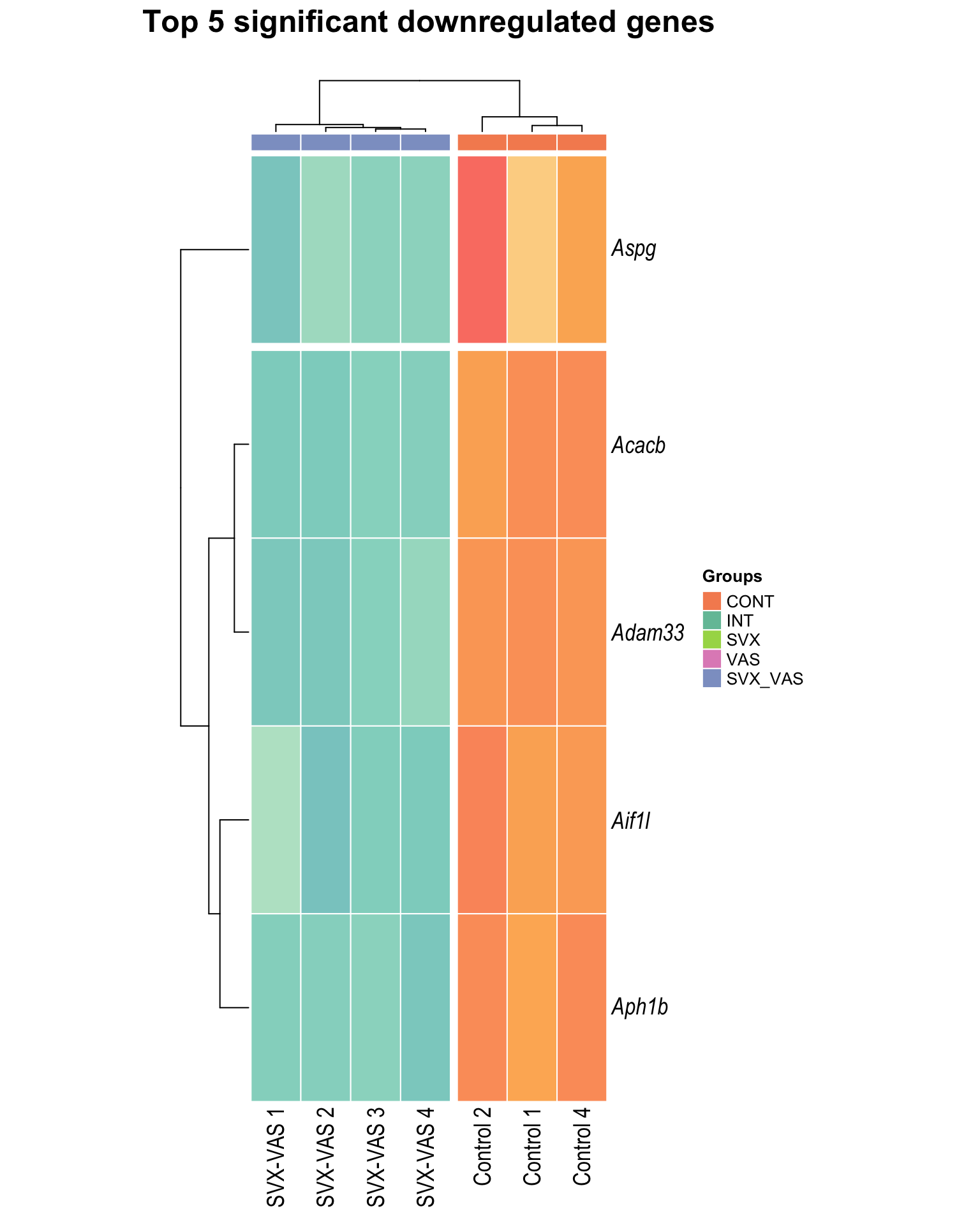
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
INT vs VAS
P-val histogram
| | | 0% | |…………………………………………………….| 100% [hist_6]
.pl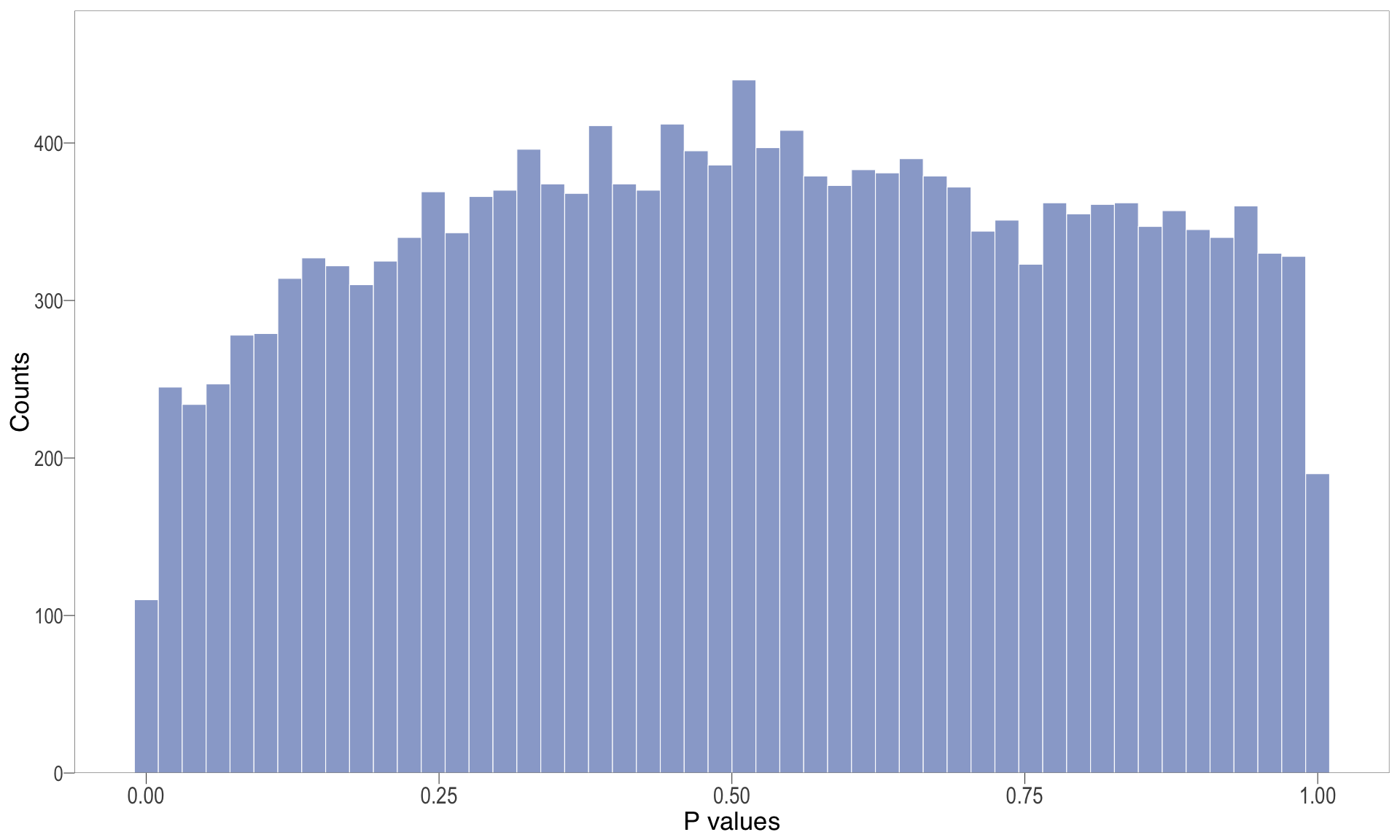
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
MA plot
| | | 0% | |………………………………………………………| 100% [ma_6]
.pl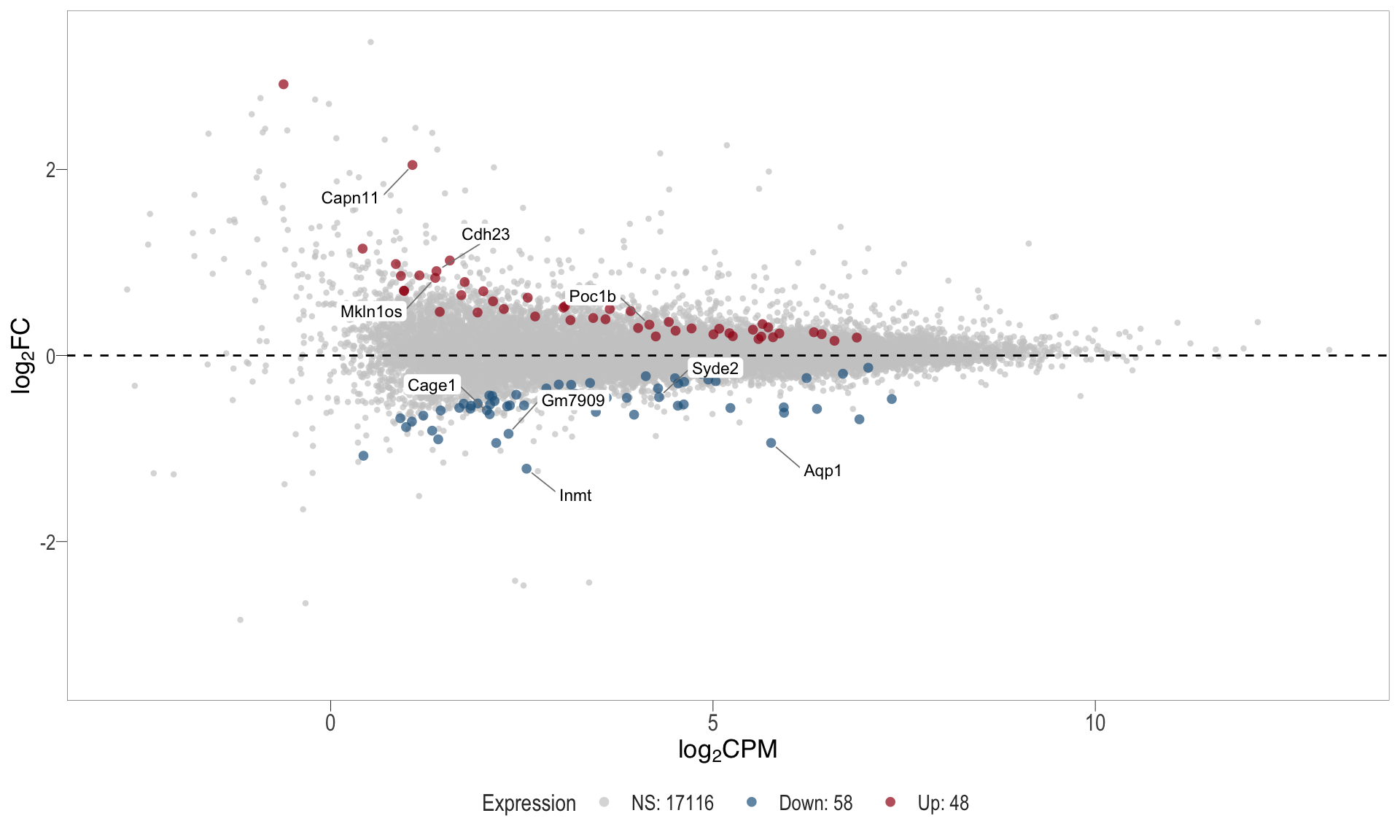
Volcano plot
| | | 0% | |……………………………………………………..| 100% [vol_6]
.pl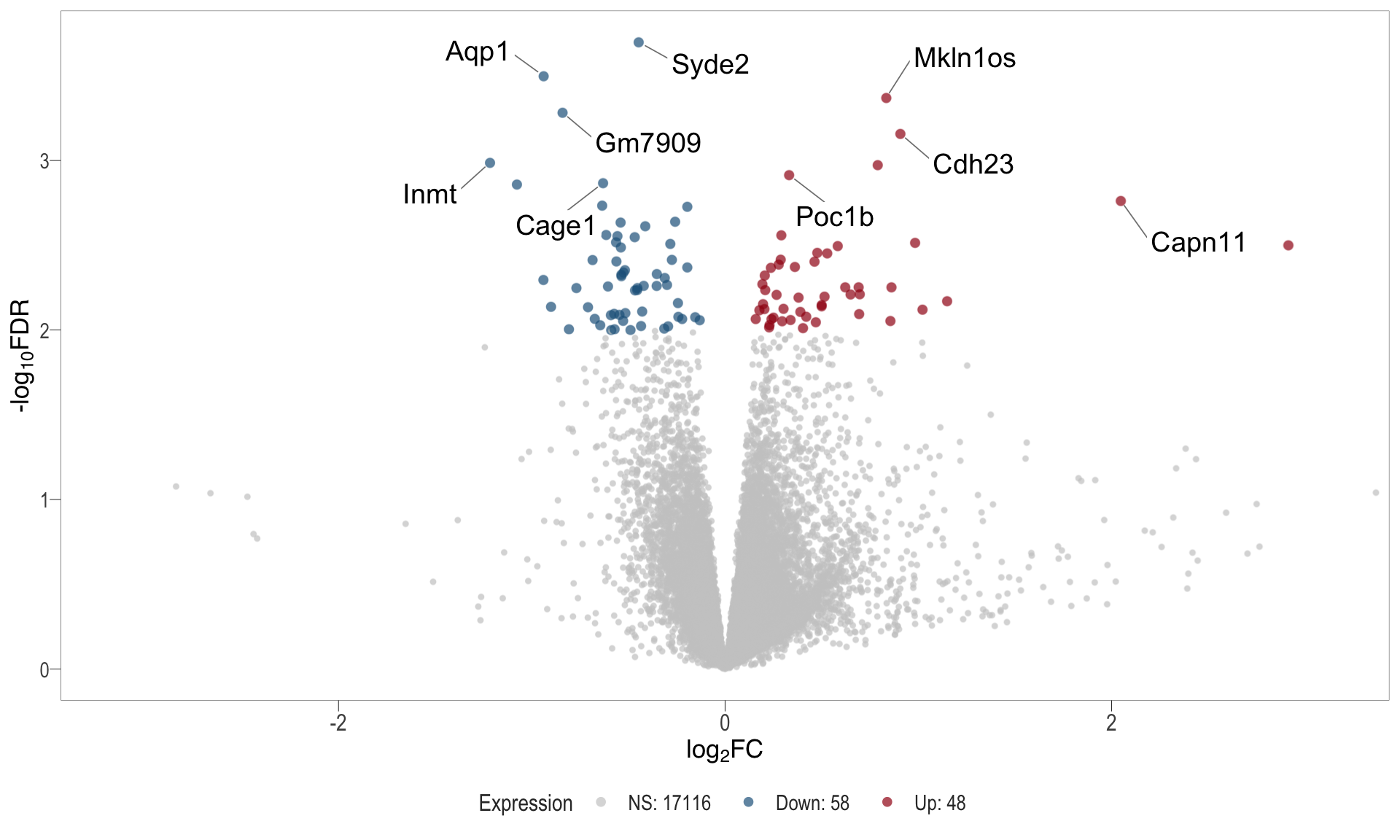
Top upregulated
| | | 0% | |…………………………………………………..| 100% [hmap_up6]
.pl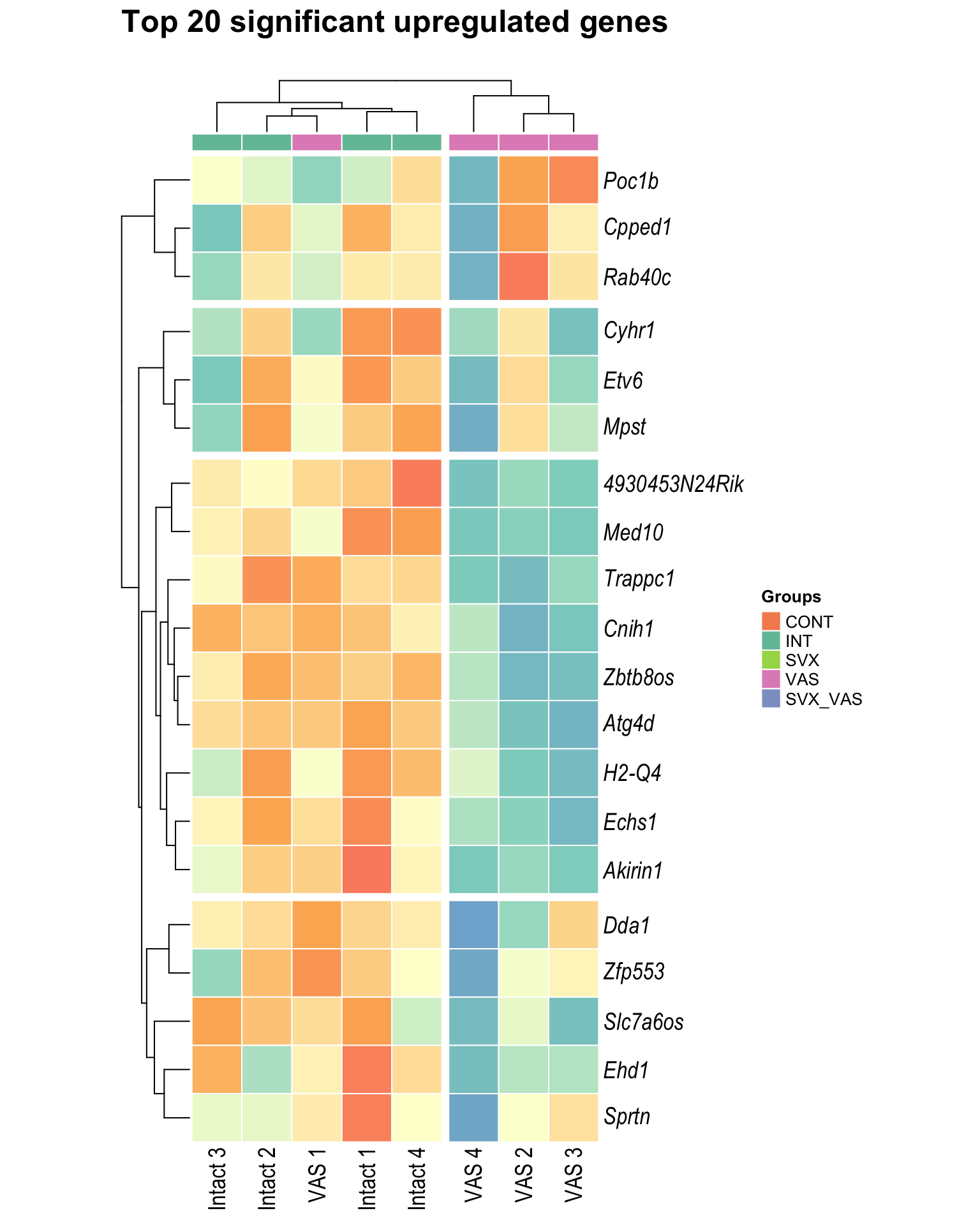
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
Top downregulated
| | | 0% | |…………………………………………………| 100% [hmap_down6]
.pl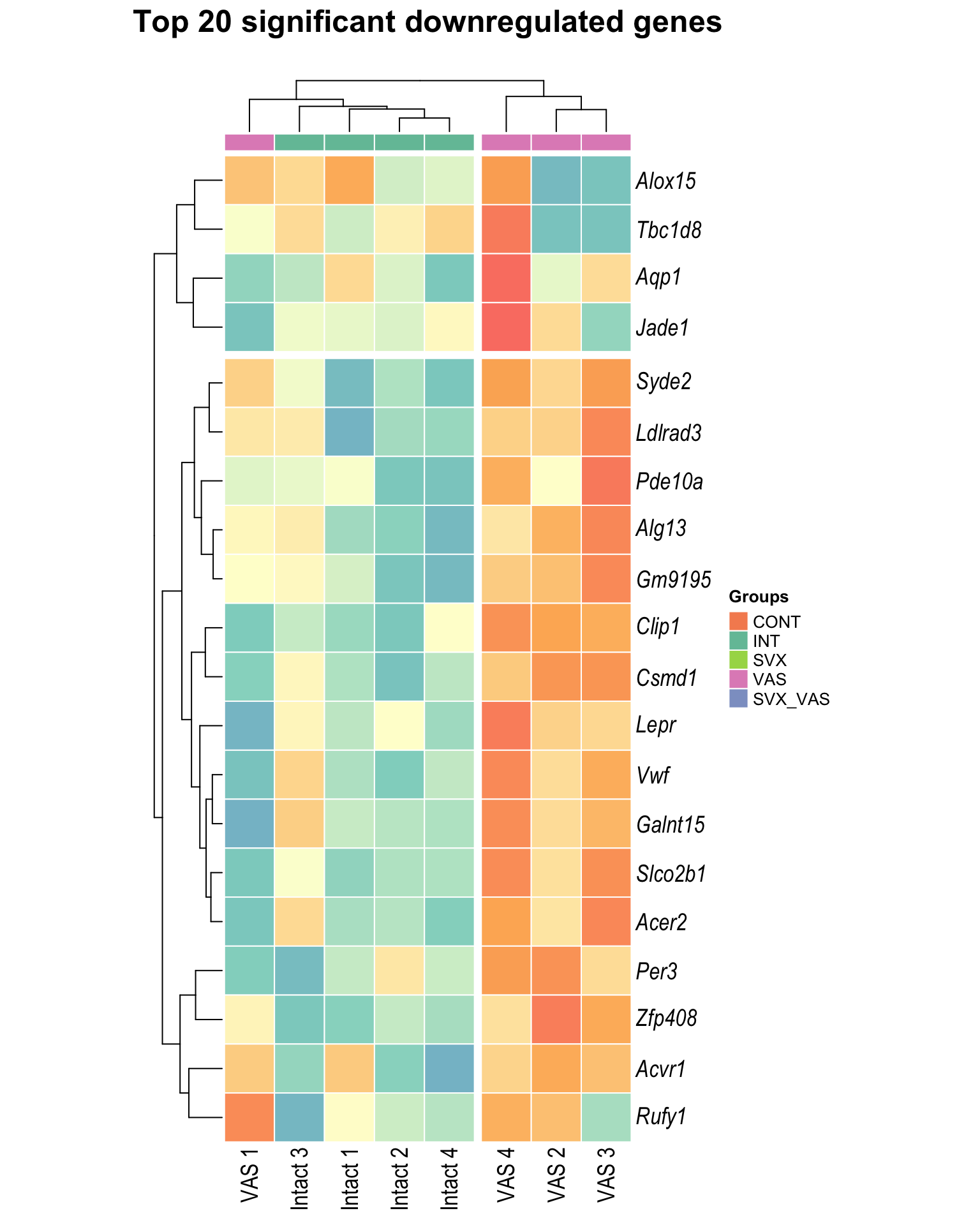
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
Combined
#create matrix with log cpm counts
logCPM <- cpm(dge, prior.count=2, log=TRUE)
rownames(logCPM) <- dge$genes$gene
#join common significant DE genes into df
common <- join_all(list(lm_sig[[1]], lm_sig[[2]], lm_sig[[3]],
lm_sig[[4]], lm_sig[[5]], lm_sig[[6]]), by = 'gene', type = 'inner')Venn diagram
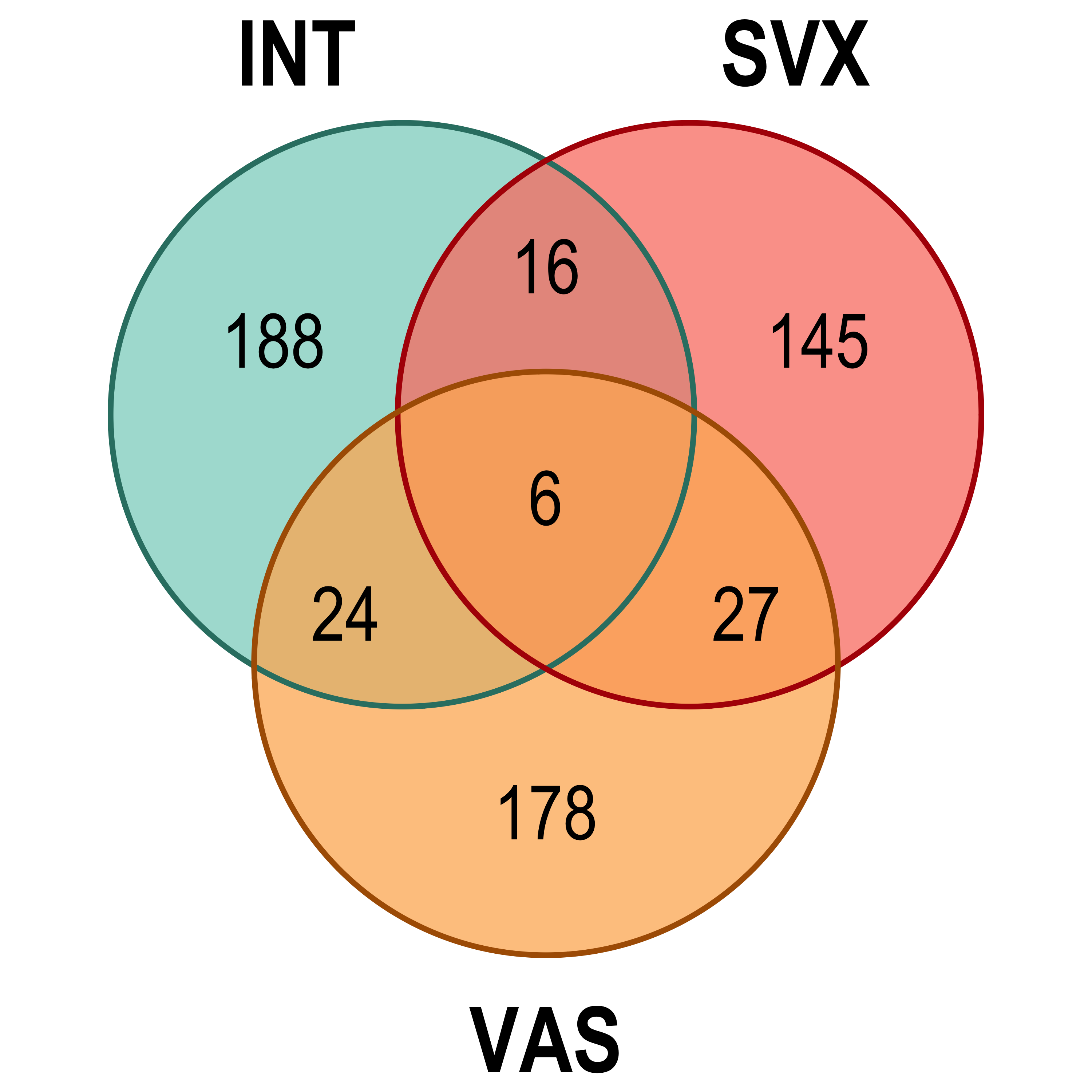

Heatmap
logCPM_combined <- cpm(dge, prior.count=3, log=TRUE)
rownames(logCPM_combined) <- dge$genes$gene_name
#join common significant DE genes into df
common <- join_all(list(lm_sig[[2]],lm_sig[[3]], lm_sig[[4]]), by = 'gene', type = 'inner')
#merge the log cpm counts with the top 30 common de genes
logCPM_combined <- logCPM_combined[common$gene_name[1:nrow(common)],]
logCPM_combined <- logCPM_combined[,c(4,5,6,7,16,17,18,19,8,9,10,11,12,13,14,15)]
#df for heatmap annotation of sample type
anno_combined <- factor(dge$samples$group, levels = c("INT", "VAS", "SVX", "SVX_VAS")) %>% as.data.frame()
anno_combined <- anno_combined[c(4,5,6,7,16,17,18,19,8,9,10,11,12,13,14,15),]%>% as.data.frame()
colnames(anno_combined) <- "Groups"
heat_combined <- ComplexHeatmap::pheatmap(
mat = logCPM_combined,
color = colorRampPalette(rev(c("#FB8072","#FDB462","#ffffd5","#8DD3C7","#80B1D3")))(300),
scale = "row",
cluster_cols = F,
border_color = "white",
gaps_col = c(4,8,12),
cutree_cols = 5,
cutree_rows = 6,
treeheight_row = 40,
treeheight_col = 30,
show_colnames = T,
clustering_distance_rows = "euclidean",
# main = paste0("Top ", nrow(logCPM_combined), " significant DEGs"),
legend = T,
heatmap_legend_param = list(title = "Expression\nZ-score",
direction= "vertical",
merge_legend = T,
legend_direction = "vertical",
legend_width = unit(5, "cm")),
# annotation = T,
annotation_legend = T,
annotation_col = anno_combined,
annotation_colors = list("Groups" = groupColour),
annotation_names_col = T,
# annotation_legend_param = list(direction = "horizontal"),
fontfamily = "Arial Narrow",
fontsize = 12,
fontsize_col = 12,
fontsize_number = 12,
fontsize_row = 12,
labels_row = as.expression(lapply(rownames(logCPM_combined), function(a) bquote(italic(.(a)))))
)
draw(heat_combined, merge_legend = T, heatmap_legend_side = "right",
annotation_legend_side = "right")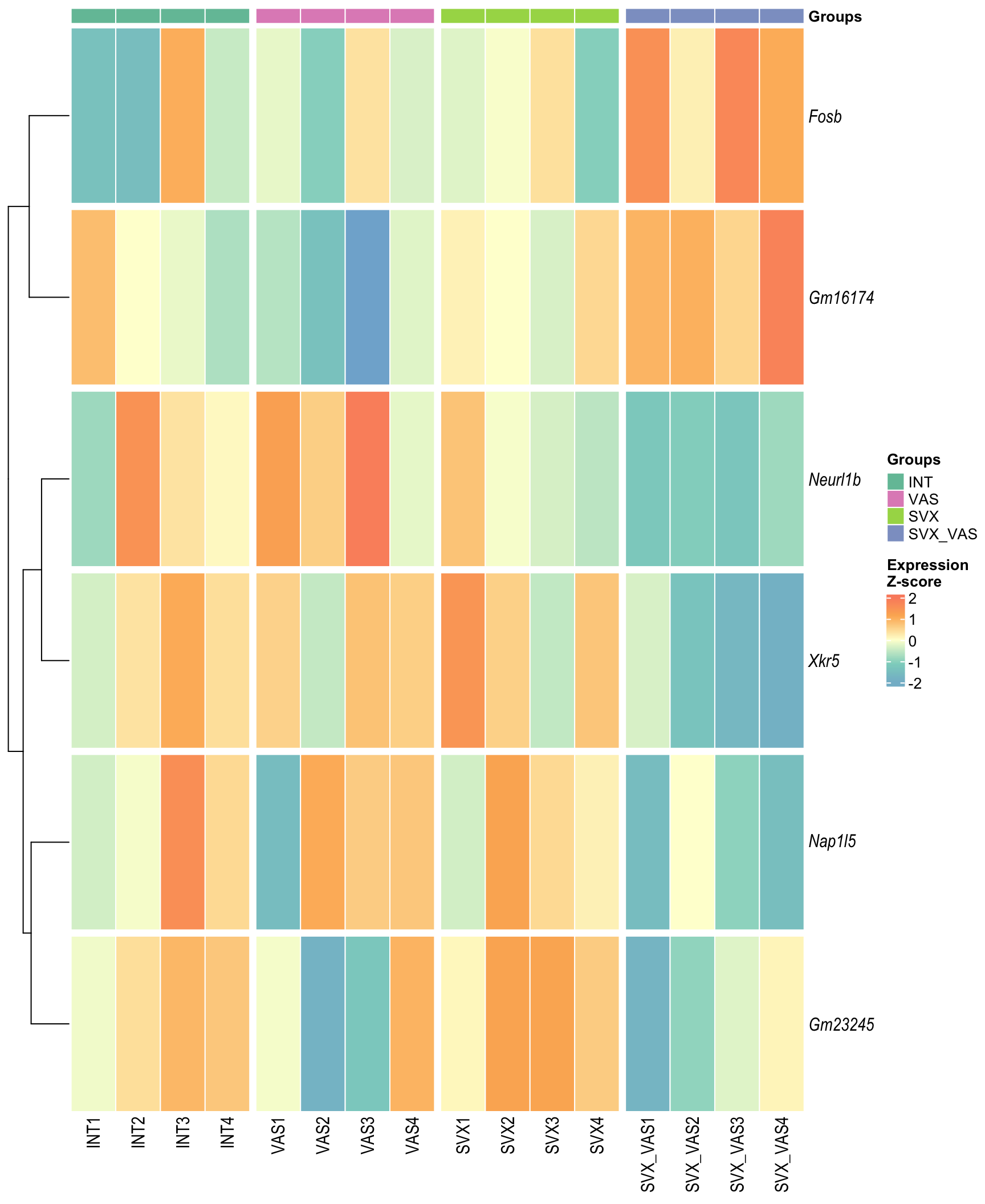
| Version | Author | Date |
|---|---|---|
| d71eeb4 | Ha Tran | 2024-10-16 |
if (savePlots == T) {
svg(filename = here::here("2_plots/2_DE/heat_combined.svg"),width = 8,height = 12)
draw(heat_combined, merge_legend = T, heatmap_legend_side = "bottom",
annotation_legend_side = "bottom")
dev.off()
}quartz_off_screen
2 Export Data
The following are exported:
de_genes_all.xlsx - This spreadsheet contains all DE genes.
de_genes_sig.xlsx - This spreadsheet contains only significant DE genes.
# export toptable for Dexter rewrite
#
lapply(lm_all, function(x) x %>% dplyr::select(c("gene_name", "logFC", "AveExpr", "P.Value", "adj.P.Val", "description", "entrezid","expression"))) %>% writexl::write_xlsx(x = ., here::here("3_output/deg_all_new.xlsx"))
lapply(lm_sig, function(x) x %>% dplyr::select(c("gene_name", "logFC", "AveExpr", "P.Value", "adj.P.Val", "description", "entrezid","expression"))) %>% writexl::write_xlsx(x = ., here::here("3_output/deg_sig_new.xlsx"))
# save RDS object for enrichment analysis
saveRDS(object = comp, file = here::here("0_data/RDS_objects/comp.rds"))
saveRDS(object = lm, file = here::here("0_data/RDS_objects/lm.rds"))
saveRDS(object = lm_all, file = here::here("0_data/RDS_objects/lm_all.rds"))
saveRDS(object = lm_sig, file = here::here("0_data/RDS_objects/lm_sig.rds"))
writexl::write_xlsx(lapply(lm_sig[c(2,3,4)], function(x) {
degs <- x %>% remove_rownames() %>% dplyr::select(c("gene_name", "gene_biotype", "description", "logFC","AveExpr","t","P.Value","adj.P.Val", "gene", "entrezid"))
degs$description <- gsub("\\[.*?\\]", "", degs$description)
return(degs)
}),path = here::here("3_output/DEGs.xlsx"))
t <- lapply(lm_sig[c(2,3,4)], function(x) {
degs <- x %>% remove_rownames() %>% dplyr::select(c("gene_name", "gene_biotype", "description", "logFC","AveExpr","t","P.Value","adj.P.Val", "gene", "entrezid"))
degs$description <- gsub("\\[.*?\\]", "", degs$description)
return(degs)
})
sessionInfo()R version 4.4.1 (2024-06-14)
Platform: aarch64-apple-darwin20
Running under: macOS 26.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Australia/Adelaide
tzcode source: internal
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] knitr_1.50 ggrastr_1.0.2 pandoc_0.2.0
[4] Glimma_2.16.0 edgeR_4.4.2 limma_3.62.2
[7] viridis_0.6.5 viridisLite_0.4.2 ggrepel_0.9.6
[10] ggpubr_0.6.1 ggplotify_0.1.3 extrafont_0.19
[13] patchwork_1.3.2 DT_0.34.0 VennDiagram_1.7.3
[16] futile.logger_1.4.3 pheatmap_1.0.13 cowplot_1.2.0
[19] pander_0.6.6 kableExtra_1.4.0 plyr_1.8.9
[22] scales_1.4.0 ComplexHeatmap_2.22.0 lubridate_1.9.4
[25] forcats_1.0.0 stringr_1.5.2 purrr_1.1.0
[28] tidyr_1.3.1 ggplot2_4.0.0 tidyverse_2.0.0
[31] reshape2_1.4.4 tibble_3.3.0 readr_2.1.5
[34] magrittr_2.0.4 dplyr_1.1.4 readxl_1.4.5
loaded via a namespace (and not attached):
[1] RColorBrewer_1.1-3 rstudioapi_0.17.1
[3] jsonlite_2.0.0 shape_1.4.6.1
[5] magick_2.9.0 ggbeeswarm_0.7.2
[7] farver_2.1.2 rmarkdown_2.29
[9] ragg_1.5.0 zlibbioc_1.52.0
[11] GlobalOptions_0.1.2 fs_1.6.6
[13] vctrs_0.6.5 Cairo_1.6-5
[15] rstatix_0.7.2 S4Arrays_1.6.0
[17] htmltools_0.5.8.1 lambda.r_1.2.4
[19] broom_1.0.10 cellranger_1.1.0
[21] SparseArray_1.6.2 Formula_1.2-5
[23] gridGraphics_0.5-1 sass_0.4.10
[25] bslib_0.9.0 htmlwidgets_1.6.4
[27] futile.options_1.0.1 cachem_1.1.0
[29] whisker_0.4.1 lifecycle_1.0.4
[31] iterators_1.0.14 pkgconfig_2.0.3
[33] Matrix_1.7-4 R6_2.6.1
[35] fastmap_1.2.0 MatrixGenerics_1.18.1
[37] GenomeInfoDbData_1.2.13 clue_0.3-66
[39] digest_0.6.37 colorspace_2.1-1
[41] S4Vectors_0.44.0 DESeq2_1.46.0
[43] rprojroot_2.1.1 crosstalk_1.2.2
[45] textshaping_1.0.3 GenomicRanges_1.58.0
[47] labeling_0.4.3 timechange_0.3.0
[49] httr_1.4.7 abind_1.4-8
[51] compiler_4.4.1 here_1.0.2
[53] withr_3.0.2 doParallel_1.0.17
[55] S7_0.2.0 backports_1.5.0
[57] BiocParallel_1.40.2 carData_3.0-5
[59] Rttf2pt1_1.3.12 ggsignif_0.6.4
[61] DelayedArray_0.32.0 rappdirs_0.3.3
[63] rjson_0.2.23 tools_4.4.1
[65] vipor_0.4.7 beeswarm_0.4.0
[67] httpuv_1.6.16 extrafontdb_1.0
[69] glue_1.8.0 promises_1.3.3
[71] cluster_2.1.8.1 generics_0.1.4
[73] gtable_0.3.6 tzdb_0.5.0
[75] hms_1.1.3 XVector_0.46.0
[77] xml2_1.4.0 car_3.1-3
[79] BiocGenerics_0.52.0 foreach_1.5.2
[81] pillar_1.11.1 yulab.utils_0.2.1
[83] later_1.4.4 circlize_0.4.16
[85] lattice_0.22-7 tidyselect_1.2.1
[87] locfit_1.5-9.12 git2r_0.36.2
[89] gridExtra_2.3 IRanges_2.40.1
[91] SummarizedExperiment_1.36.0 svglite_2.2.1
[93] stats4_4.4.1 xfun_0.53
[95] Biobase_2.66.0 statmod_1.5.0
[97] matrixStats_1.5.0 stringi_1.8.7
[99] UCSC.utils_1.2.0 workflowr_1.7.2
[101] yaml_2.3.10 evaluate_1.0.5
[103] codetools_0.2-20 cli_3.6.5
[105] systemfonts_1.2.3 jquerylib_0.1.4
[107] Rcpp_1.1.0 GenomeInfoDb_1.42.3
[109] png_0.1-8 parallel_4.4.1
[111] writexl_1.5.4 crayon_1.5.3
[113] GetoptLong_1.0.5 rlang_1.1.6
[115] formatR_1.14