GO Analysis
Ha M. Tran
22/08/2021
Last updated: 2023-09-16
Checks: 7 0
Knit directory:
Mouse_endometrial_transcriptome_2023/1_analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(12345) was run prior to running the
code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 18c6463. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rproj.user/
Untracked files:
Untracked: .gitignore
Untracked: 0_data/RDS_objects/DT.rds
Unstaged changes:
Modified: 0_data/RDS_objects/dge.rds
Modified: 0_data/RDS_objects/enrichGO.rds
Modified: 0_data/RDS_objects/enrichGO_sig.rds
Modified: 0_data/RDS_objects/fc.rds
Modified: 0_data/RDS_objects/lfc.rds
Modified: 0_data/RDS_objects/lmTreat.rds
Modified: 0_data/RDS_objects/lmTreat_all.rds
Modified: 0_data/RDS_objects/lmTreat_sig.rds
Modified: 0_data/RDS_objects/pub.rds
Deleted: 1_analysis/mmu04060.pv.png
Deleted: 1_analysis/mmu04151.pv.png
Deleted: 1_analysis/mmu04270.pv.png
Deleted: 1_analysis/mmu04510.pv.png
Deleted: 1_analysis/mmu04640.pv.png
Deleted: 1_analysis/mmu04670.pv.png
Modified: 2_plots/de/pval_1.05.svg
Modified: 2_plots/de/pval_1.1.svg
Modified: 2_plots/de/pval_1.5.svg
Modified: 2_plots/go/bp_dot_1.05.svg
Modified: 2_plots/go/bp_dot_1.5.svg
Modified: 2_plots/go/cc_dot_1.05.svg
Modified: 2_plots/go/cc_dot_1.5.svg
Modified: 2_plots/go/mf_dot_1.5.svg
Modified: 2_plots/go/upset_1.05.svg
Modified: 2_plots/go/upset_1.1.svg
Modified: 2_plots/go/upset_1.5.svg
Modified: 2_plots/ipa/Cell-To-Cell Signaling.svg
Modified: 2_plots/ipa/diseaseAndFunction.svg
Modified: 2_plots/ipa/pathways.svg
Modified: 2_plots/kegg/kegg_dot_1.05.svg
Modified: 2_plots/kegg/kegg_dot_1.1.svg
Modified: 2_plots/kegg/kegg_dot_1.5.svg
Modified: 2_plots/kegg/upset_kegg_1.05.svg
Modified: 2_plots/kegg/upset_kegg_1.1.svg
Modified: 2_plots/kegg/upset_kegg_1.5.svg
Modified: 2_plots/qc/PCA_IntvsCont.svg
Modified: 2_plots/qc/counts_after_filtering_3_3.svg
Modified: 2_plots/qc/counts_before_after_filtering_3_3.svg
Modified: 2_plots/qc/counts_before_filtering.svg
Modified: 2_plots/qc/library_size.svg
Modified: 2_plots/reactome/react_dot_1.05.svg
Modified: 2_plots/reactome/react_dot_1.1.svg
Modified: 2_plots/reactome/react_dot_1.5.svg
Modified: 2_plots/reactome/upset_react_1.05.svg
Modified: 2_plots/reactome/upset_react_1.1.svg
Modified: 2_plots/reactome/upset_react_1.5.svg
Modified: 3_output/enrichGO_sig.xlsx
Modified: 3_output/enrichKEGG_all.xlsx
Modified: 3_output/enrichKEGG_sig.xlsx
Modified: 3_output/lmTreat_all.xlsx
Modified: 3_output/lmTreat_fc1.5_voom2_all_fdr.xlsx
Modified: 3_output/lmTreat_sig.xlsx
Modified: 3_output/reactome_all.xlsx
Modified: 3_output/reactome_sig.xlsx
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (1_analysis/go.Rmd) and HTML
(docs/go.html) files. If you’ve configured a remote Git
repository (see ?wflow_git_remote), click on the hyperlinks
in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 18c6463 | Ha Manh Tran | 2023-09-16 | Added KEGG copyright permission and changed kable |
| html | b44640a | Ha Manh Tran | 2023-01-23 | Build site. |
| Rmd | eb10bef | Ha Manh Tran | 2023-01-22 | workflowr::wflow_publish(here::here("1_analysis/*.Rmd")) |
| html | 8c9178d | tranmanhha135 | 2023-01-21 | added png |
| html | d578f46 | Ha Manh Tran | 2023-01-21 | Build site. |
| html | 159f352 | tranmanhha135 | 2023-01-21 | adding for pathview |
| Rmd | 4d51a4e | tranmanhha135 | 2023-01-20 | test png |
| html | 4d51a4e | tranmanhha135 | 2023-01-20 | test png |
| html | 691cf34 | Ha Manh Tran | 2023-01-20 | Build site. |
| Rmd | 7f6bab2 | Ha Manh Tran | 2023-01-20 | workflowr::wflow_publish(here::here("1_analysis/*.Rmd")) |
| Rmd | b6cf190 | tranmanhha135 | 2023-01-19 | quick commit |
| Rmd | 3119fad | tranmanhha135 | 2022-11-05 | build website |
| html | 3119fad | tranmanhha135 | 2022-11-05 | build website |
Data Setup
# working with data
library(dplyr)
library(magrittr)
library(readr)
library(tibble)
library(reshape2)
library(tidyverse)
# Visualisation:
library(kableExtra)
library(ggplot2)
library(grid)
library(DT)
# Custom ggplot
library(gridExtra)
library(ggbiplot)
library(ggrepel)
# Bioconductor packages:
library(edgeR)
library(limma)
library(Glimma)
library(clusterProfiler)
library(org.Mm.eg.db)
library(enrichplot)
theme_set(theme_minimal())
pub <- readRDS(here::here("0_data/RDS_objects/pub.rds"))
DT <- readRDS(here::here("0_data/RDS_objects/DT.rds"))Import DGElist Data
DGElist object containing the raw feature count, sample metadata, and gene metadata, created in the Set Up stage.
# load DGElist previously created in the set up
dge <- readRDS(here::here("0_data/RDS_objects/dge.rds"))
fc <- readRDS(here::here("0_data/RDS_objects/fc.rds"))
lfc <- readRDS(here::here("0_data/RDS_objects/lfc.rds"))
lmTreat <- readRDS(here::here("0_data/RDS_objects/lmTreat.rds"))
lmTreat_sig <- readRDS(here::here("0_data/RDS_objects/lmTreat_sig.rds"))GO Analysis
goSummaries is a package created by Dr Stephen Pederson
for filtering GO terms based on ontology level.
# circumvent rerunning of lengthy analysis.
enrichGO <- readRDS(here::here("0_data/RDS_objects/enrichGO.rds"))
enrichGO_sig <- readRDS(here::here("0_data/RDS_objects/enrichGO_sig.rds"))# download go summaries and set the minimum ontology level
goSummaries <- url("https://uofabioinformaticshub.github.io/summaries2GO/data/goSummaries.RDS") %>%
readRDS()
minPath <- 3
enrichGO=list()
enrichGO_sig <- list()
for (i in 1:length(fc)) {
x <- fc[i] %>% as.character()
# find enriched GO terms
enrichGO[[x]] <- clusterProfiler::enrichGO(
gene = lmTreat_sig[[x]]$entrezid,
OrgDb = org.Mm.eg.db,
keyType = "ENTREZID",
ont = "ALL",
pAdjustMethod = "fdr",
pvalueCutoff = 0.05
)
# bind to goSummaries to elminate go terms with ontology levels 1 and 2.
enrichGO_sig[[x]] <- enrichGO[[x]] %>%
clusterProfiler::setReadable(OrgDb = org.Mm.eg.db, keyType = "auto")
enrichGO_sig[[x]] <- enrichGO_sig[[x]] %>%
as.data.frame() %>%
rownames_to_column("id") %>%
left_join(goSummaries) %>%
dplyr::filter(shortest_path >= minPath) %>%
column_to_rownames("id")
# adjust go results, separate compound column, add FDR column, adjust the GeneRatio column
enrichGO_sig[[x]] <- enrichGO_sig[[x]] %>%
separate(col = BgRatio, sep = "/", into = c("Total", "Universe")) %>%
dplyr::mutate(
logFDR = -log(p.adjust, 10),
GeneRatio = Count / as.numeric(Total)) %>%
dplyr::select(c("Description", "ontology", "GeneRatio", "pvalue", "p.adjust", "logFDR", "qvalue", "geneID", "Count"))
# at the beginnning of a word (after 35 characters), add a newline. shorten the y axis for dot plot
# enrichGO_sig[[x]]$Description <- sub(pattern = "(.{1,35})(?:$| )",
# replacement = "\\1\n",
# x = enrichGO_sig[[x]]$Description)
# # remove the additional newline at the end of the string
# enrichGO_sig[[x]]$Description <- sub(pattern = "\n$",
# replacement = "",
# x = enrichGO_sig[[x]]$Description)
}FC=1.05
# display the top 30 most sig
enrichGO_sig[[1]] %>%
dplyr::mutate_if(is.numeric, funs(as.character(signif(.,3)))) %>%
DT(., caption = "Significantly enriched GO terms") # kable(caption = "Significantly enriched GO terms") %>%
# kable_styling(bootstrap_options = c("striped", "hover")) %>%
# scroll_box(height = "600px")Visualisation
bp_dot=list()
mf_dot=list()
cc_dot=list()
upset=list()
for (i in 1:length(fc)) {
x <- fc[i] %>% as.character()
# extract the enriched GO terms from each ontology
bp <- enrichGO_sig[[x]] %>% dplyr::filter(ontology == "BP")
mf <- enrichGO_sig[[x]] %>% dplyr::filter(ontology == "MF")
cc <- enrichGO_sig[[x]] %>% dplyr::filter(ontology == "CC")
# bp dot plot, save
bp_dot[[x]] <- ggplot(bp[1:15, ]) +
geom_point(aes(x = GeneRatio, y = reorder(Description, GeneRatio), colour = logFDR, size = Count)) +
scale_color_gradient(low = "dodgerblue3", high = "firebrick3", limits = c(0, NA)) +
scale_size(range = c(.5,3)) +
ggtitle("Biological Process") +
ylab(label = "") +
xlab(label = "Gene Ratio") +
labs(color = expression("-log"[10] * "FDR"), size = "Gene Counts")
ggsave(filename = paste0("bp_dot_", fc[i], ".svg"), plot = bp_dot[[x]] + pub, path = here::here("2_plots/go/"),
width = 200, height = 120, units = "mm")
# mf dot plot, save
mf_dot[[x]] <- ggplot(mf[1:15, ]) +
geom_point(aes(x = GeneRatio, y = reorder(Description, GeneRatio), colour = logFDR, size = Count)) +
scale_color_gradient(low = "dodgerblue3", high = "firebrick3", limits = c(0, NA)) +
scale_size(range = c(.5,3)) +
ggtitle("Molecular Function") +
ylab(label = "") +
xlab(label = "Gene Ratio") +
labs(color = expression("-log"[10] * "FDR"), size = "Gene Counts")
ggsave(filename = paste0("mf_dot_", fc[i], ".svg"), plot = mf_dot[[x]] + pub, path = here::here("2_plots/go/"),
width = 200, height = 120, units = "mm")
# cc dot plot, save
cc_dot[[x]] <- ggplot(cc[1:15, ]) +
geom_point(aes(x = GeneRatio, y = reorder(Description, GeneRatio), colour = logFDR, size = Count)) +
scale_color_gradient(low = "dodgerblue3", high = "firebrick3", limits = c(0, NA)) +
scale_size(range = c(.5,3)) +
ggtitle("Cellular Components") +
ylab(label = "") +
xlab(label = "Gene Ratio") +
labs(color = expression("-log"[10] * "FDR"), size = "Gene Counts")
ggsave(filename = paste0("cc_dot_", fc[i], ".svg"), plot = cc_dot[[x]] + pub, path = here::here("2_plots/go/"),
width = 200, height = 120, units = "mm")
upset[[x]] <- upsetplot(x = enrichGO[[x]], 10)
ggsave(filename = paste0("upset_", fc[i], ".svg"), plot = upset[[x]], path = here::here("2_plots/go/"), width = 250, height = 166, units = "mm")
}Biological Process
bp_dot[[1]] 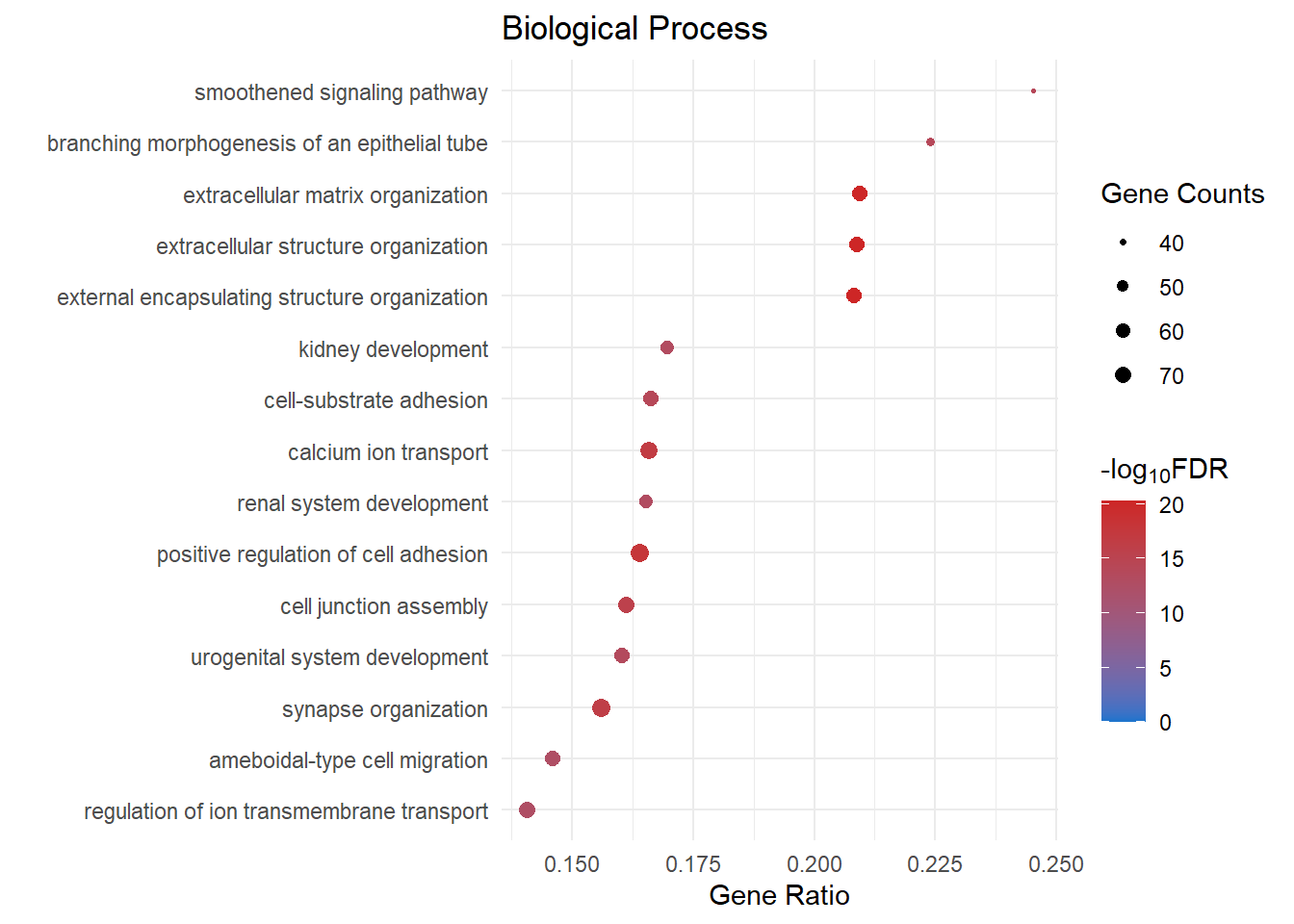
Molecular Function
mf_dot[[1]]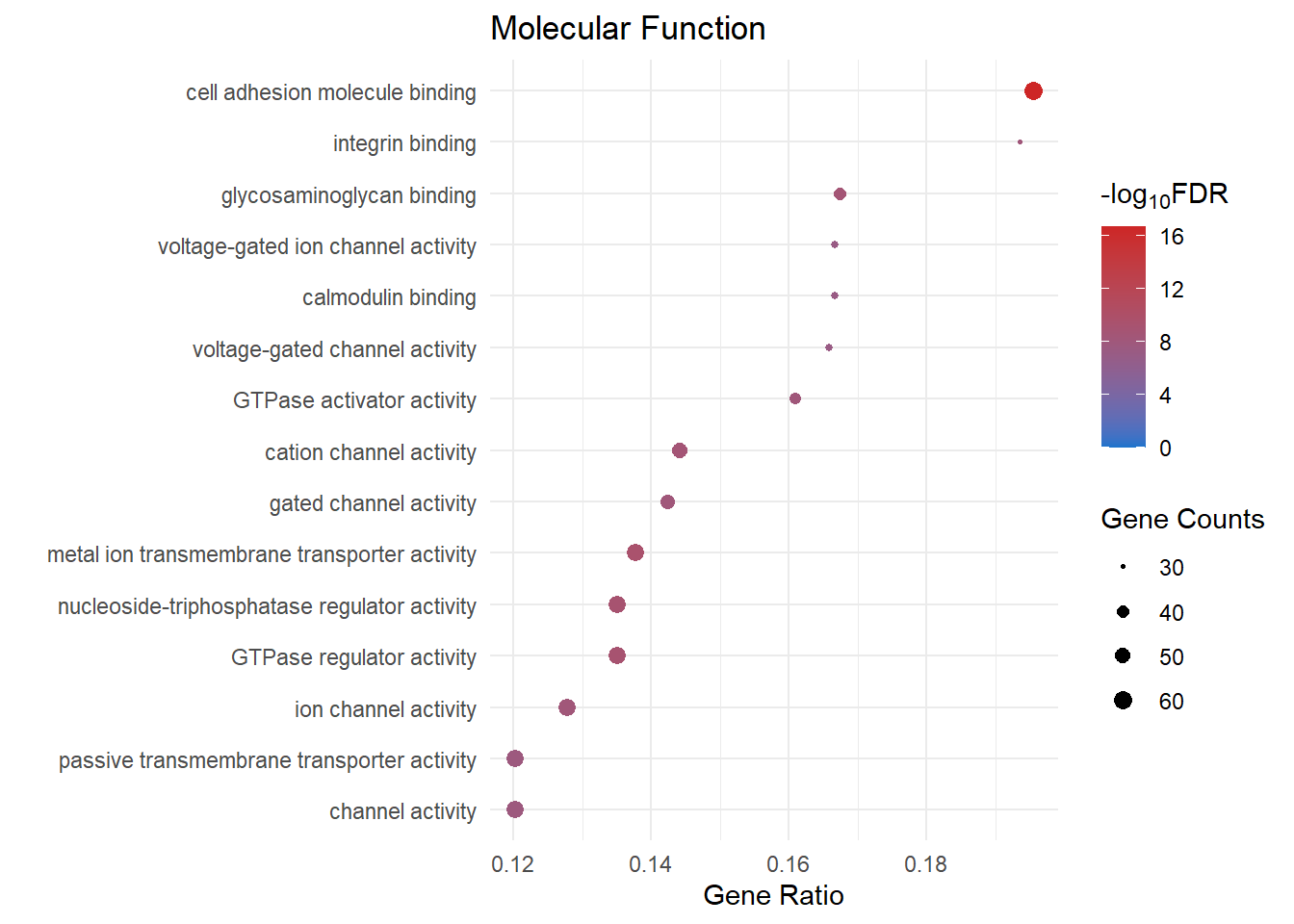
Cellular Components
cc_dot[[1]]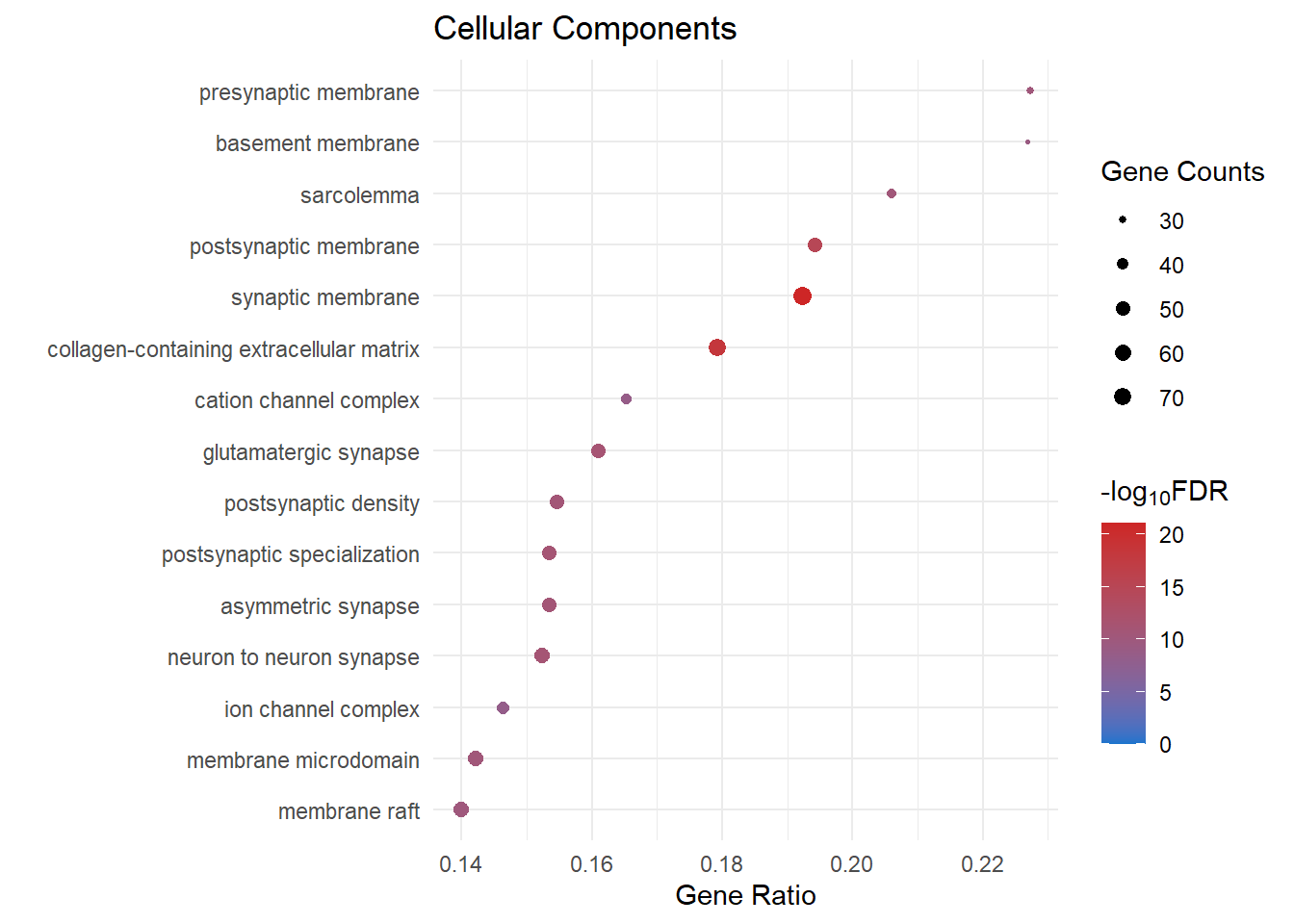
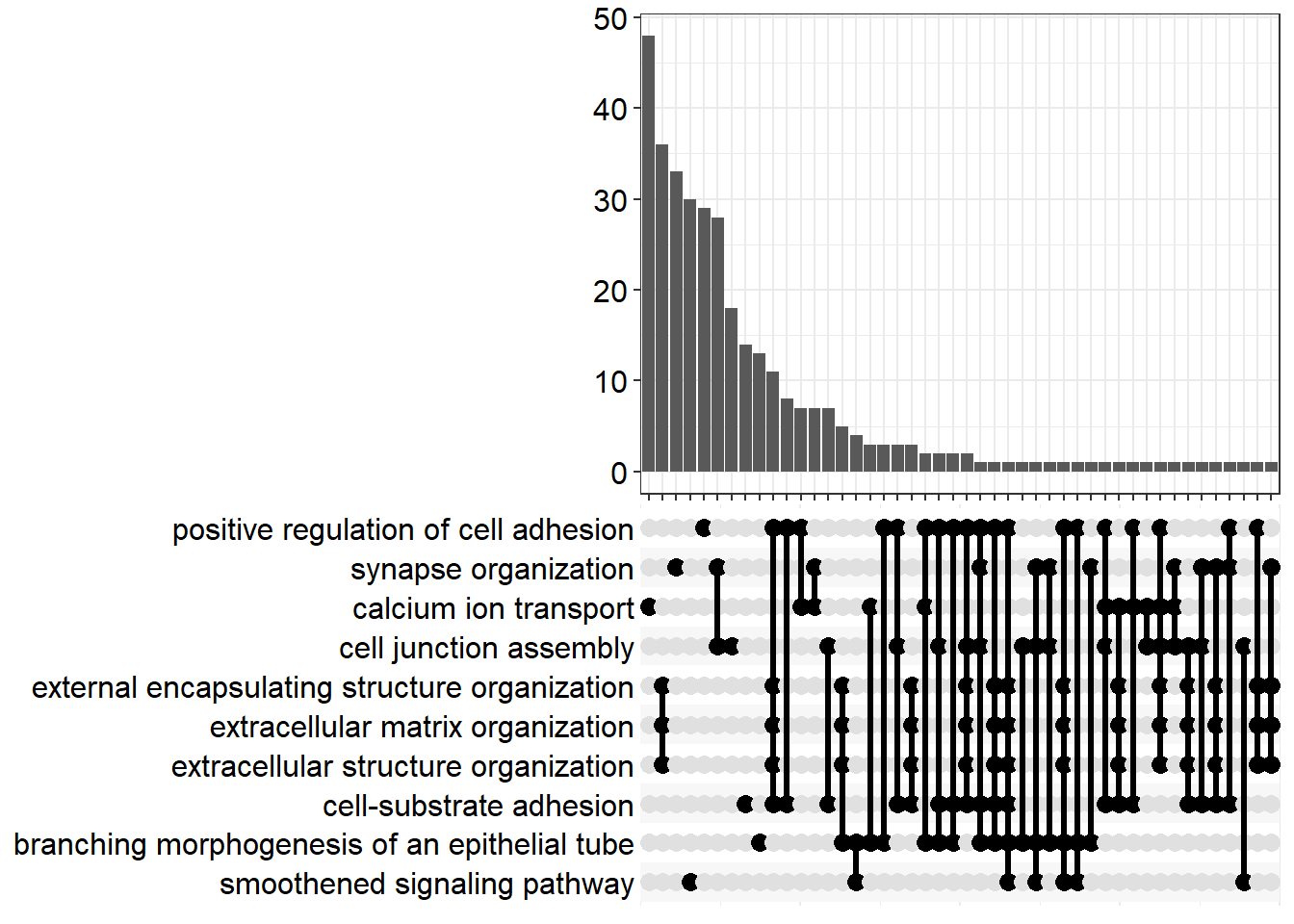
FC=1.1
# display the top 30 most sig
enrichGO_sig[[2]] %>%
dplyr::mutate_if(is.numeric, funs(as.character(signif(.,3)))) %>%
DT(.,caption = "Significantly enriched GO terms") # kable(caption = "Significantly enriched GO terms") %>%
# kable_styling(bootstrap_options = c("striped", "hover")) %>%
# scroll_box(height = "600px")Visualisation
Biological Process
bp_dot[[2]]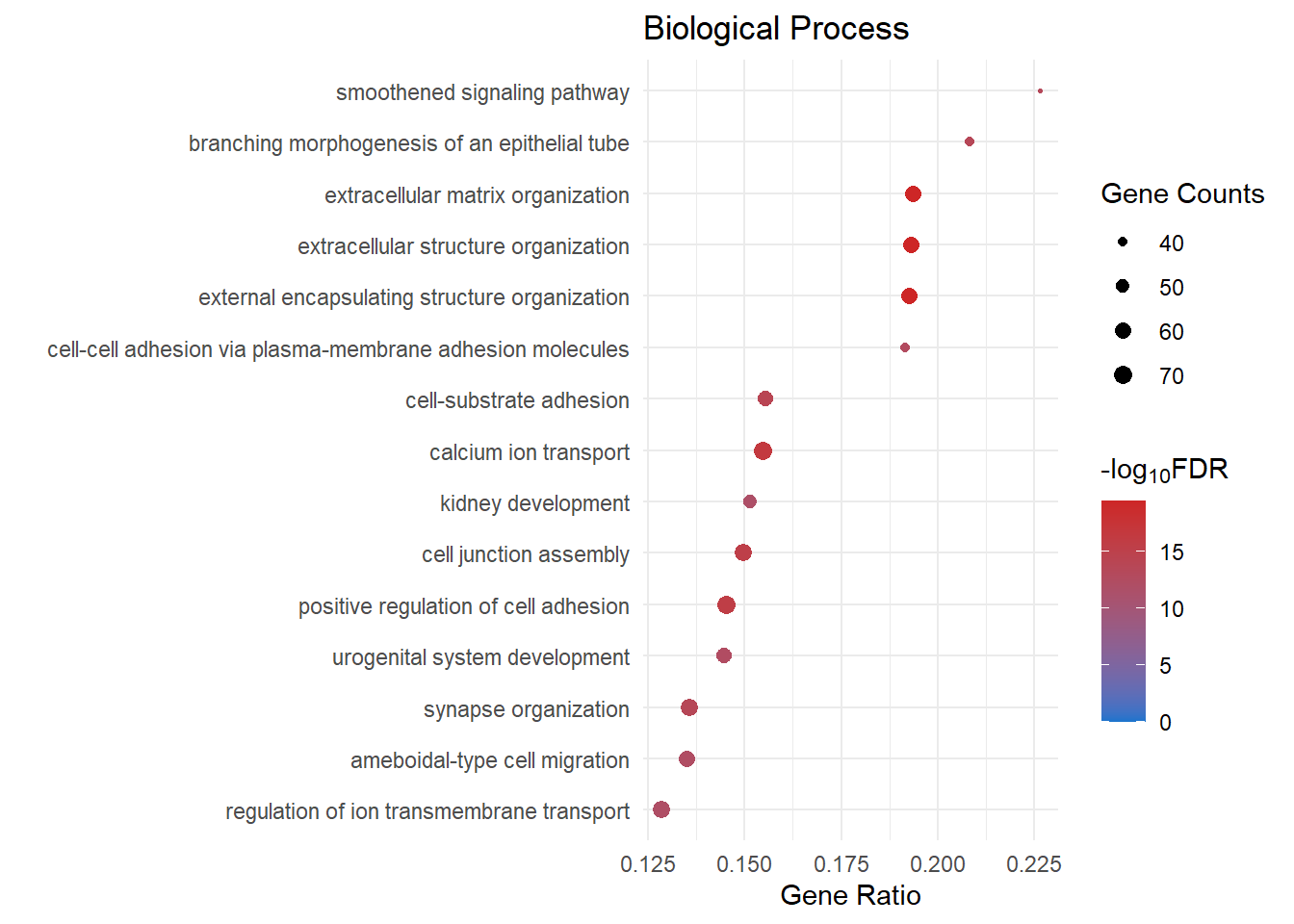
Molecular Function
mf_dot[[2]]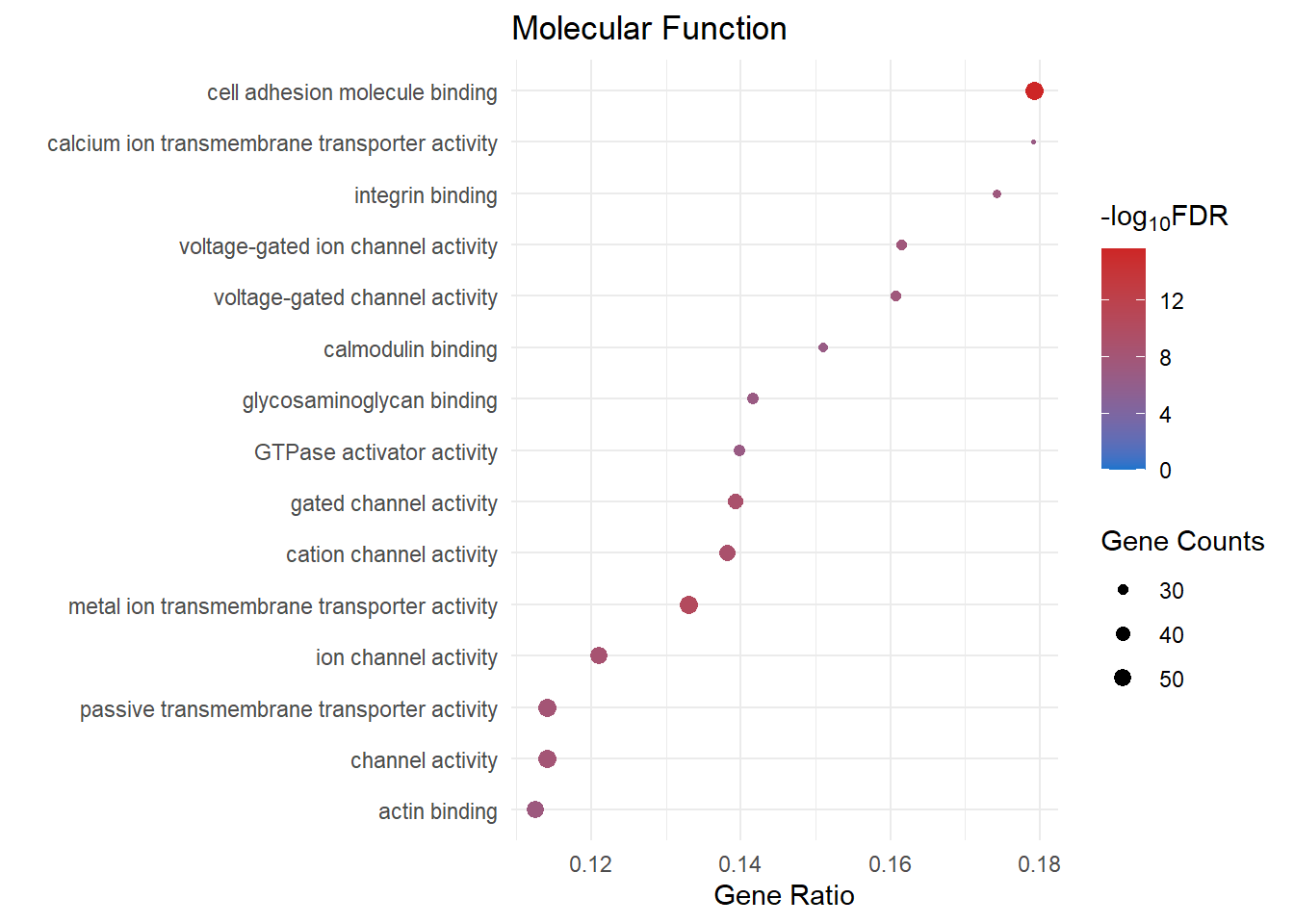
Cellular Components
cc_dot[[2]]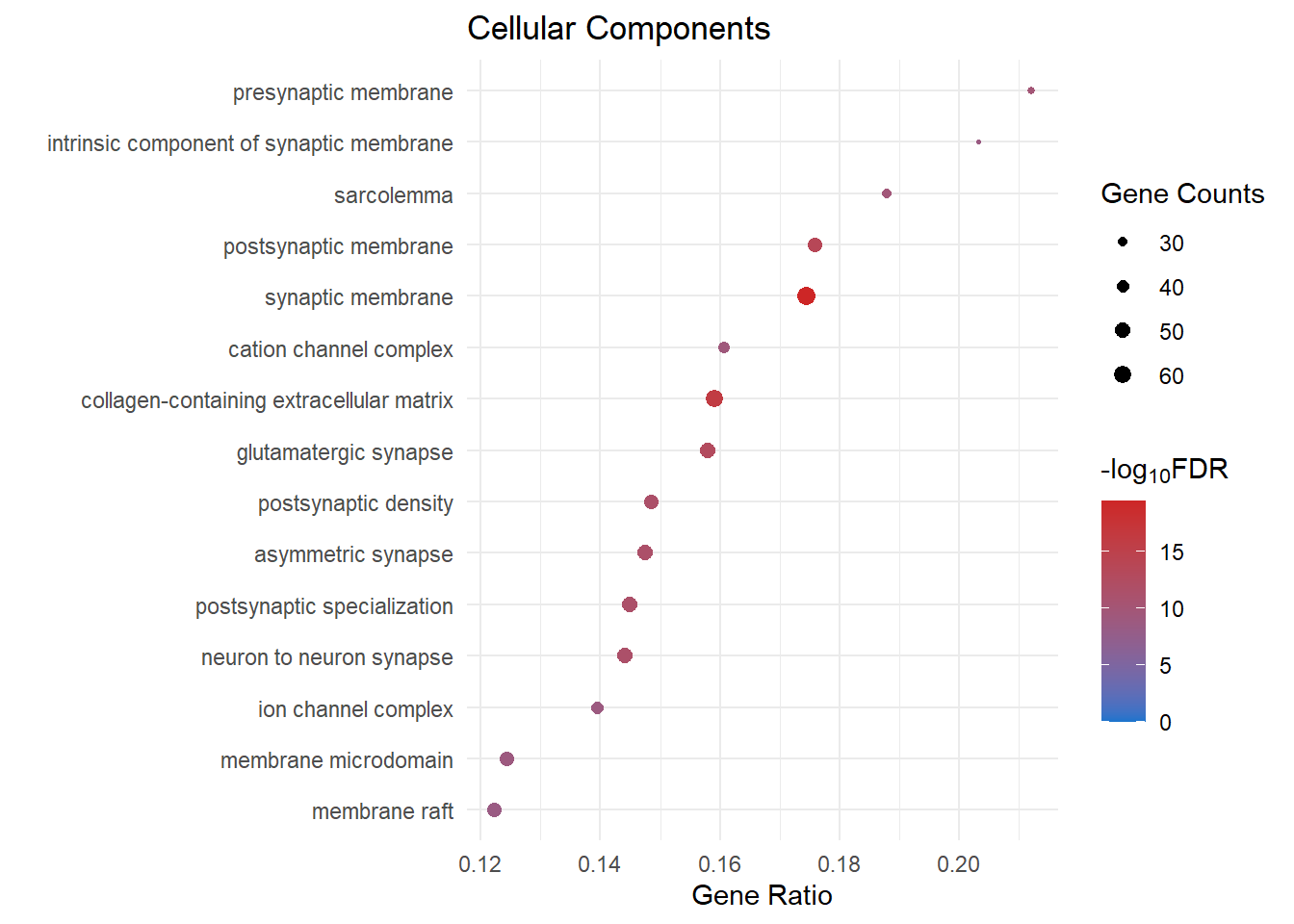
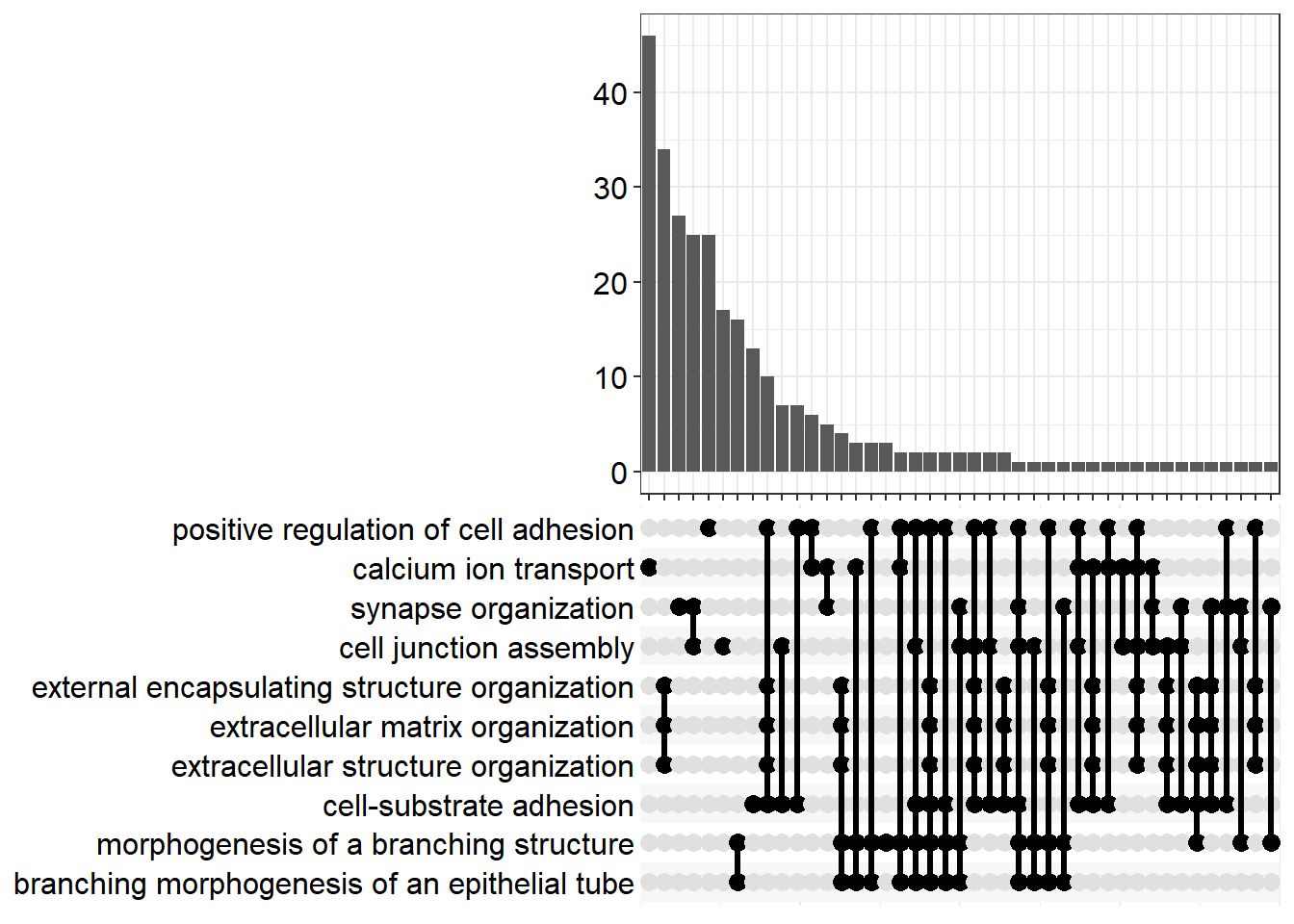
FC=1.5
# display the top 30 most sig
enrichGO_sig[[3]] %>%
dplyr::mutate_if(is.numeric, funs(as.character(signif(.,3)))) %>%
DT(.,caption = "Significantly enriched GO terms") # kable(caption = "Significantly enriched GO terms") %>%
# kable_styling(bootstrap_options = c("striped", "hover")) %>%
# scroll_box(height = "600px")Visualisation
Biological Process
bp_dot[[3]]
Molecular Function
mf_dot[[3]]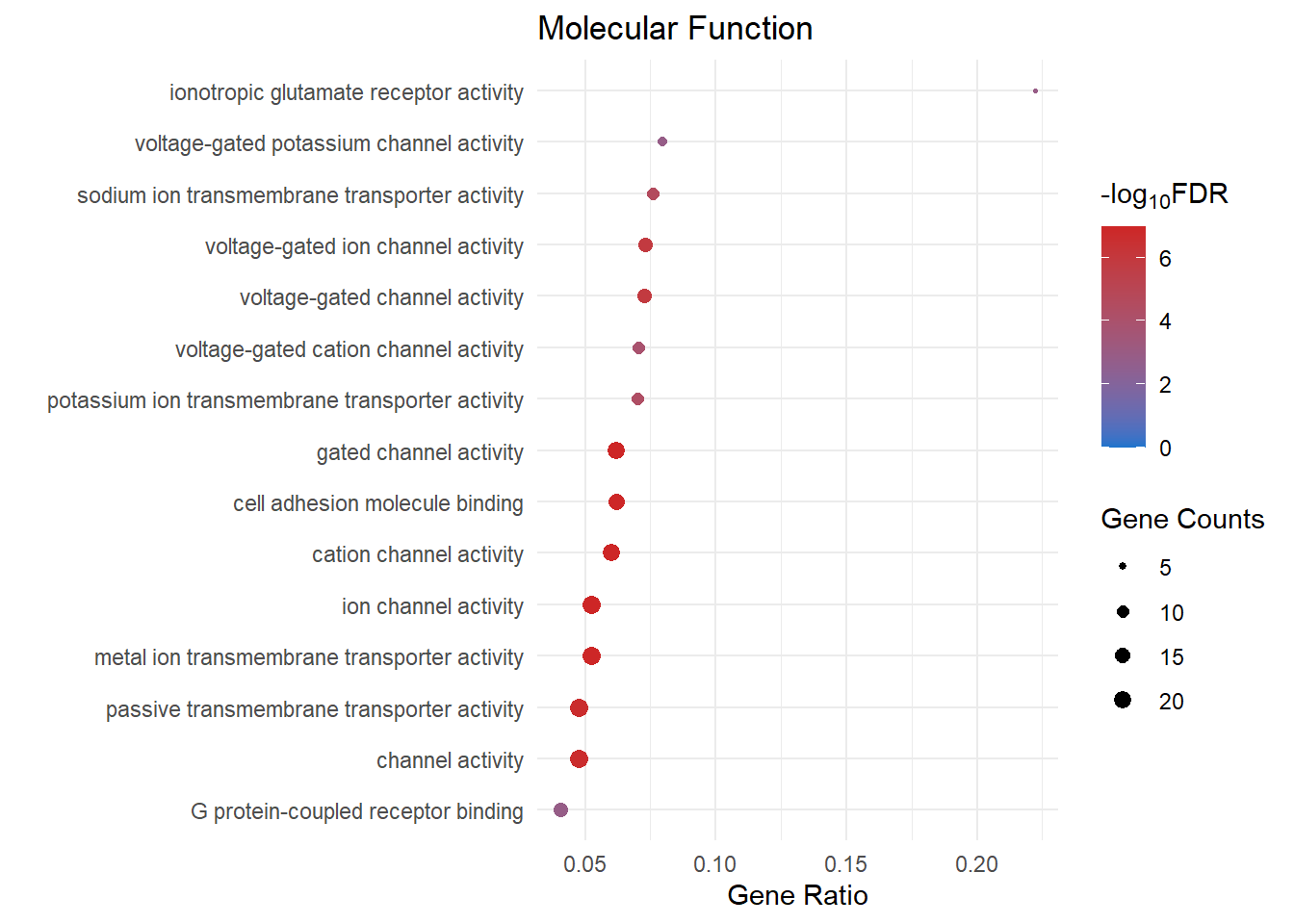
Cellular Components
cc_dot[[3]]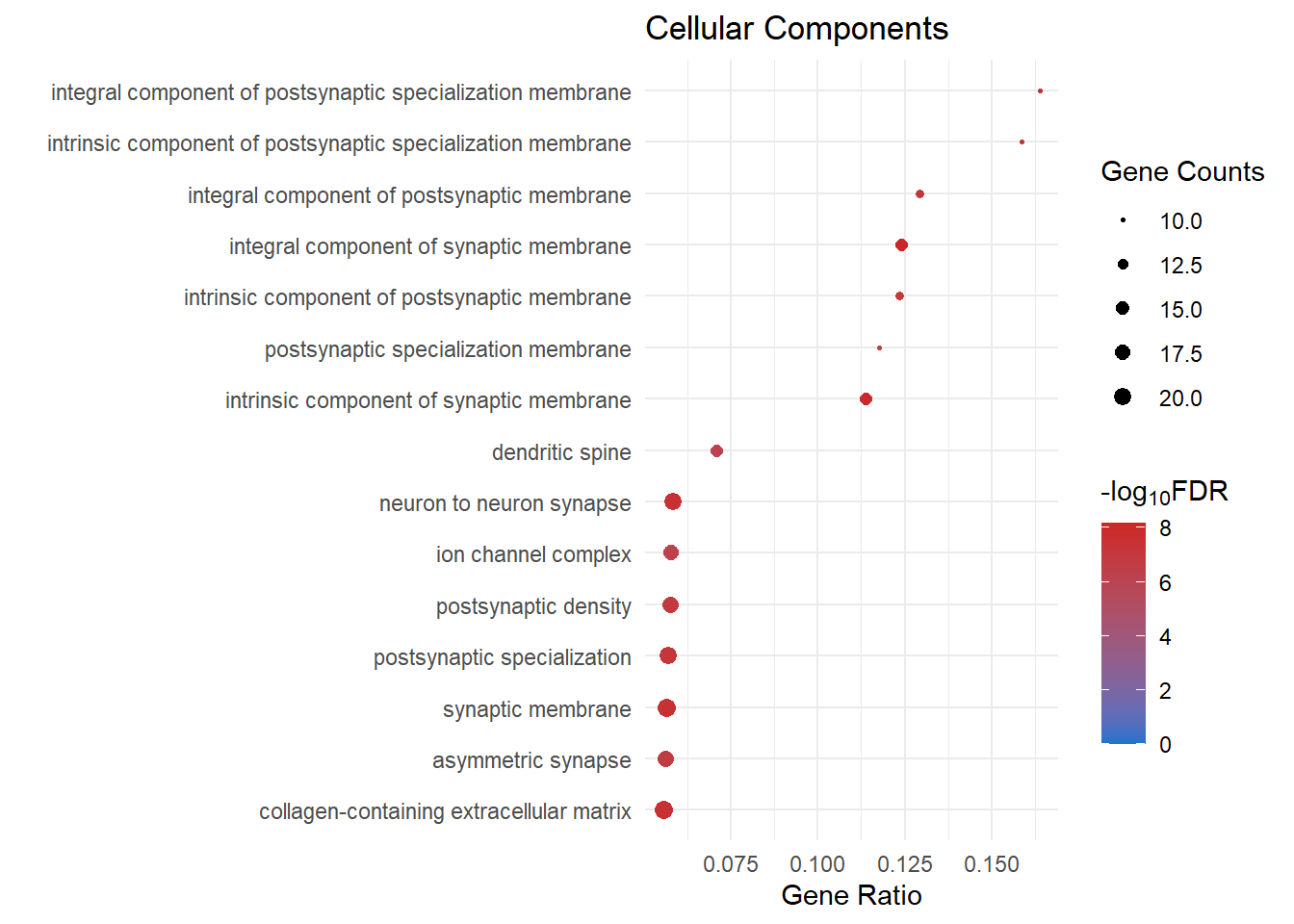
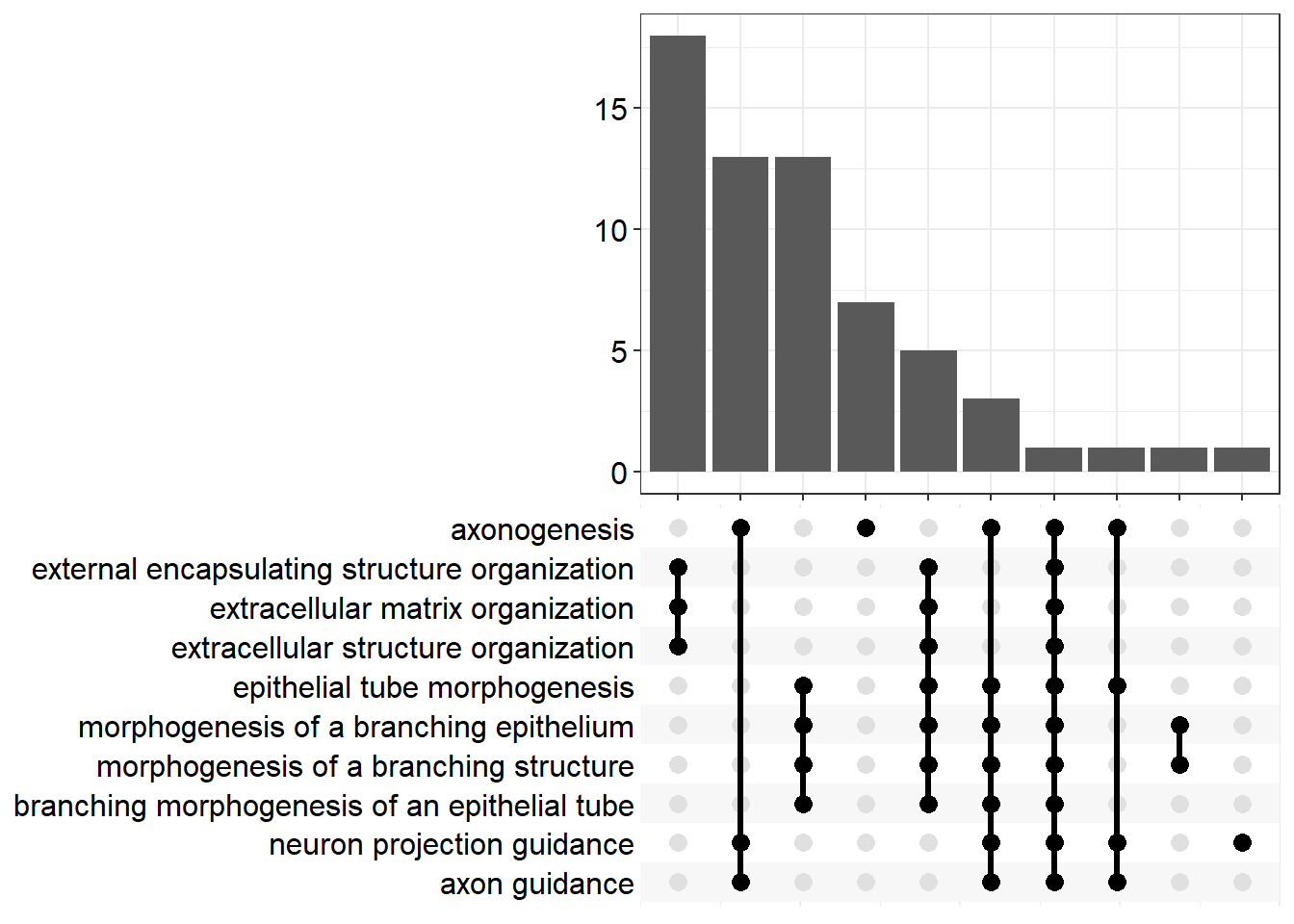
Export Data
# save to excel
writexl::write_xlsx(x = enrichGO_sig, here::here("3_output/enrichGO_sig.xlsx"))
saveRDS(object = enrichGO_sig,file = here::here("0_data/RDS_objects/enrichGO_sig.rds"))
saveRDS(object = enrichGO,file = here::here("0_data/RDS_objects/enrichGO.rds"))
sessionInfo()R version 4.3.1 (2023-06-16 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19045)
Matrix products: default
locale:
[1] LC_COLLATE=English_Australia.utf8 LC_CTYPE=English_Australia.utf8
[3] LC_MONETARY=English_Australia.utf8 LC_NUMERIC=C
[5] LC_TIME=English_Australia.utf8
time zone: Australia/Adelaide
tzcode source: internal
attached base packages:
[1] stats4 grid stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] enrichplot_1.20.1 org.Mm.eg.db_3.17.0 AnnotationDbi_1.62.2
[4] IRanges_2.34.1 S4Vectors_0.38.1 Biobase_2.60.0
[7] BiocGenerics_0.46.0 clusterProfiler_4.8.3 Glimma_2.10.0
[10] edgeR_3.42.4 limma_3.56.2 ggrepel_0.9.3
[13] ggbiplot_0.55 scales_1.2.1 plyr_1.8.8
[16] gridExtra_2.3 DT_0.29 kableExtra_1.3.4
[19] lubridate_1.9.2 forcats_1.0.0 stringr_1.5.0
[22] purrr_1.0.1 tidyr_1.3.0 ggplot2_3.4.3
[25] tidyverse_2.0.0 reshape2_1.4.4 tibble_3.2.1
[28] readr_2.1.4 magrittr_2.0.3 dplyr_1.1.2
loaded via a namespace (and not attached):
[1] splines_4.3.1 later_1.3.1
[3] bitops_1.0-7 ggplotify_0.1.2
[5] polyclip_1.10-4 lifecycle_1.0.3
[7] rprojroot_2.0.3 lattice_0.21-8
[9] MASS_7.3-60 crosstalk_1.2.0
[11] sass_0.4.7 rmarkdown_2.24
[13] jquerylib_0.1.4 yaml_2.3.7
[15] httpuv_1.6.11 cowplot_1.1.1
[17] DBI_1.1.3 RColorBrewer_1.1-3
[19] abind_1.4-5 zlibbioc_1.46.0
[21] rvest_1.0.3 GenomicRanges_1.52.0
[23] ggraph_2.1.0 RCurl_1.98-1.12
[25] yulab.utils_0.0.9 tweenr_2.0.2
[27] git2r_0.32.0 GenomeInfoDbData_1.2.10
[29] tidytree_0.4.5 svglite_2.1.1
[31] codetools_0.2-19 DelayedArray_0.26.7
[33] DOSE_3.26.1 xml2_1.3.5
[35] ggforce_0.4.1 tidyselect_1.2.0
[37] aplot_0.2.1 farver_2.1.1
[39] viridis_0.6.4 matrixStats_1.0.0
[41] webshot_0.5.5 jsonlite_1.8.7
[43] ellipsis_0.3.2 tidygraph_1.2.3
[45] systemfonts_1.0.4 tools_4.3.1
[47] treeio_1.24.3 ragg_1.2.5
[49] Rcpp_1.0.11 glue_1.6.2
[51] xfun_0.39 here_1.0.1
[53] DESeq2_1.40.2 qvalue_2.32.0
[55] MatrixGenerics_1.12.3 GenomeInfoDb_1.36.3
[57] withr_2.5.0 fastmap_1.1.1
[59] fansi_1.0.4 digest_0.6.33
[61] timechange_0.2.0 R6_2.5.1
[63] gridGraphics_0.5-1 textshaping_0.3.6
[65] colorspace_2.1-0 GO.db_3.17.0
[67] RSQLite_2.3.1 utf8_1.2.3
[69] generics_0.1.3 data.table_1.14.8
[71] graphlayouts_1.0.0 httr_1.4.7
[73] htmlwidgets_1.6.2 S4Arrays_1.0.6
[75] scatterpie_0.2.1 whisker_0.4.1
[77] pkgconfig_2.0.3 gtable_0.3.4
[79] blob_1.2.4 workflowr_1.7.1
[81] XVector_0.40.0 shadowtext_0.1.2
[83] htmltools_0.5.5 fgsea_1.26.0
[85] ggupset_0.3.0 png_0.1-8
[87] ggfun_0.1.3 knitr_1.44
[89] rstudioapi_0.15.0 tzdb_0.4.0
[91] nlme_3.1-163 cachem_1.0.8
[93] parallel_4.3.1 HDO.db_0.99.1
[95] pillar_1.9.0 vctrs_0.6.3
[97] promises_1.2.0.1 evaluate_0.21
[99] cli_3.6.1 locfit_1.5-9.8
[101] compiler_4.3.1 rlang_1.1.1
[103] crayon_1.5.2 labeling_0.4.3
[105] fs_1.6.3 writexl_1.4.2
[107] stringi_1.7.12 viridisLite_0.4.2
[109] BiocParallel_1.34.2 munsell_0.5.0
[111] Biostrings_2.68.1 lazyeval_0.2.2
[113] GOSemSim_2.26.1 Matrix_1.6-1
[115] hms_1.1.3 patchwork_1.1.3
[117] bit64_4.0.5 KEGGREST_1.40.0
[119] SummarizedExperiment_1.30.2 igraph_1.5.1
[121] memoise_2.0.1 bslib_0.5.1
[123] ggtree_3.8.2 fastmatch_1.1-4
[125] bit_4.0.5 downloader_0.4
[127] ape_5.7-1 gson_0.1.0